Gene Expression Profiling as a Tool to Investigate the Molecular Machinery Activated during Hippocampal Neurodegeneration Induced by Trimethyltin (TMT) Administration

Abstract
:1. Introduction
2. Different Models Used to Investigate TMT-Induced Gene Expression Profiling
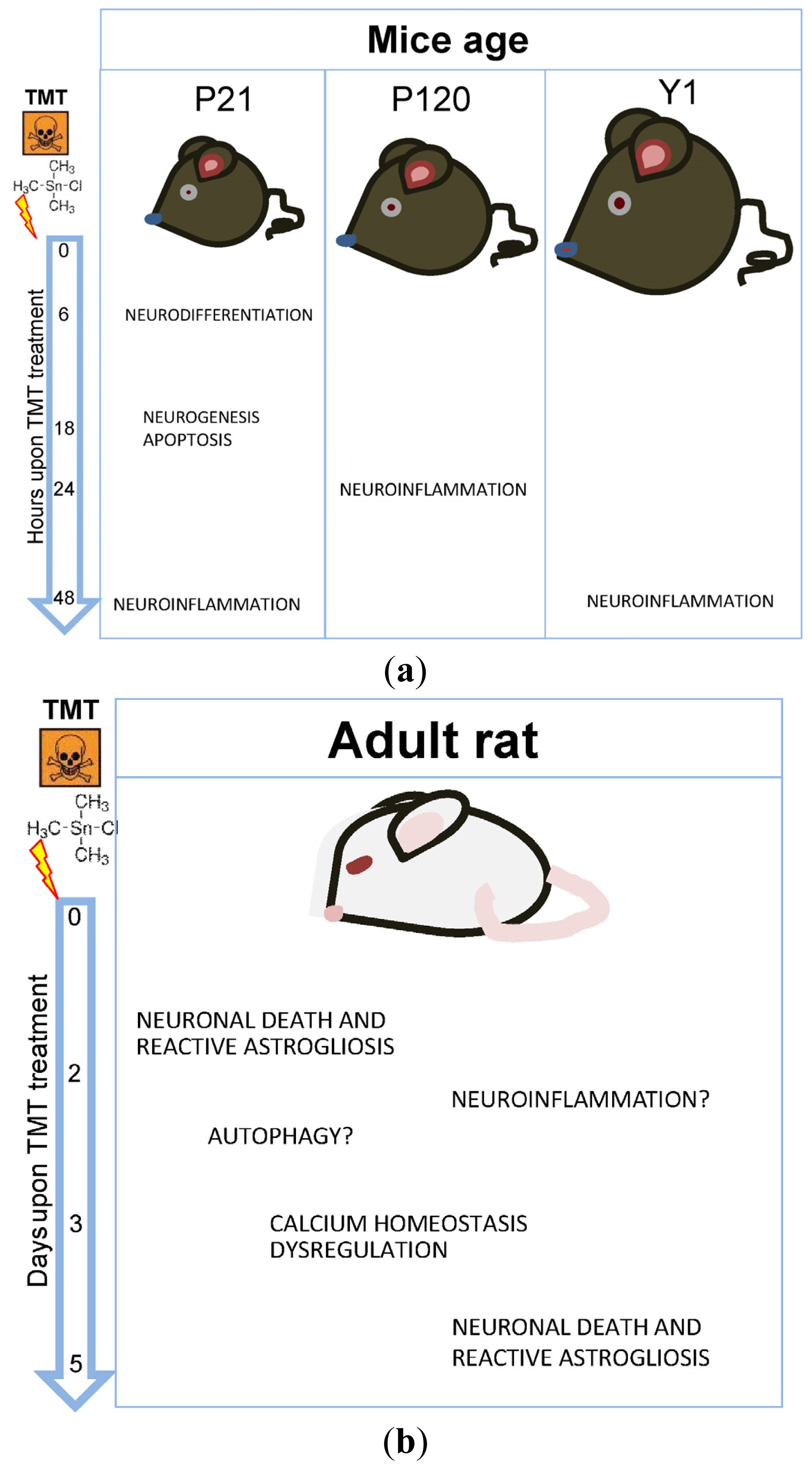
2.1. The Mouse Model
2.2. The Rat Model
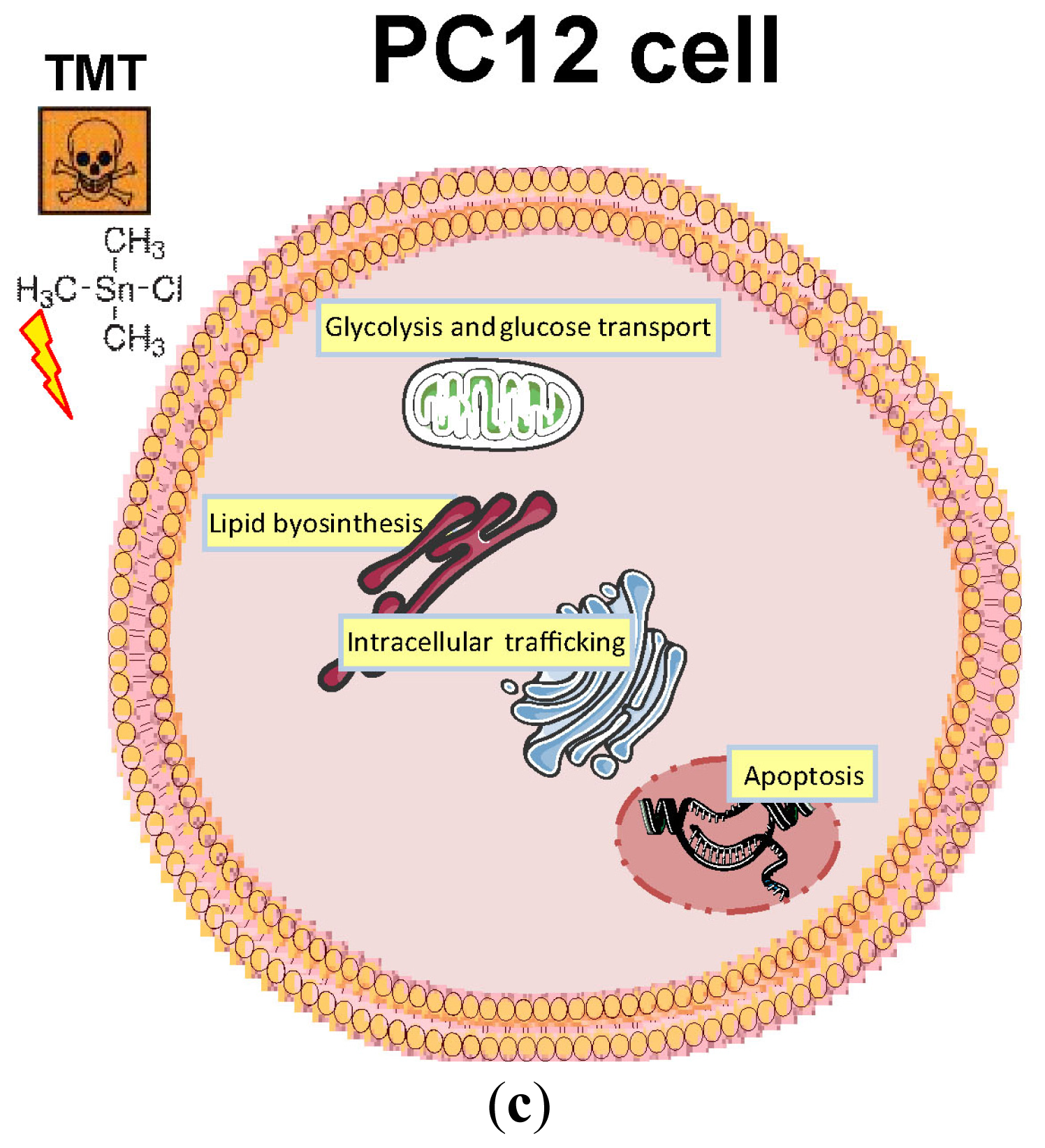
2.3. The Cell Culture Model
3. Concluding Remarks and Perspectives
Acknowledgments
Conflicts of Interest
References
- Chang, L.W.; Wenger, G.R.; McMillan, D.E.; Dyer, R.S. Species and strain comparison of acute neurotoxic effects of trimethyltin in mice and rats. Neurobehav. Toxicol. Teratol 1983, 5, 337–350. [Google Scholar]
- Chang, L.W. The neurotoxicology and pathology of organomercury, organolead, and organotin. J. Toxicol. Sci. Suppl 1990, 4, 125–151. [Google Scholar]
- Boyer, I.J. Toxicity of dibutyltin, tributyltin and other organotin compounds to humans and to experimental animals. Toxicology 1989, 55, 253–298. [Google Scholar]
- Jiang, G.B.; Zhou, Q.F.; He, B. Tin compounds and major trace metal elements in organotin poisoned patient’s urine and blood measured by gas chromatography flame photometric detector and inductively coupled plasma mass spectrometry. Bull. Environ. Contam. Toxicol 2000, 65, 277–284. [Google Scholar]
- Saary, M.J.; House, R.A. Preventable exposure to trimethyltin chloride: A case report. Occup. Med 2002, 52, 227–230. [Google Scholar]
- Yoo, C.I.; Kim, Y.; Jeong, K.S.; Sim, C.S.; Choy, N.; Kim, J.; Eum, J.B.; Nakaiama, Y.; Endo, Y.; Kim, Y.J. A case of acute organotin poisoning. J. Occup. Health 2007, 49, 305–310. [Google Scholar]
- Geloso, M.C.; Corvino, V.; Michetti, F. Trimethyltin-induced hippocampal degeneration as a tool to investigate neurodegenerative processes. Neurochem. Int 2011, 58, 729–738. [Google Scholar]
- Corvino, V.; Marchese, E.; Michetti, F.; Geloso, M.C. Neuroprotective strategies in hippocampal neurodegeneration induced by the neurotoxicant trimethyltin. Neurochem. Res 2013, 38, 240–253. [Google Scholar]
- Ogita, K.; Nishiyama, N.; Sugiyama, C.; Higuchi, K.; Yoneyama, M.; Yoneda, Y. Regeneration of granule neurons after lesioning of hippocampal dentate gyrus: Evaluation using adult mice treated with trimethyltin chloride as a model. J. Neurosci. Res 2005, 82, 609–621. [Google Scholar]
- McPherson, C.A.; Aoyama, M.; Harry, G.J. Interleukin (IL)-1 and IL-6 regulation of neural progenitor cell proliferation with hippocampal injury: Differential regulatory pathways in the subgranular zone (SGZ) of the adolescent and mature mouse brain. Brain Behav. Immun 2011, 25, 850–862. [Google Scholar]
- Corvino, V.; Geloso, M.C.; Cavallo, V.; Guadagni, E.; Passalacqua, R.; Florenzano, F.; Giannetti, S.; Molinari, M.; Michetti, F. Enhanced neurogenesis during trimethyltin-induced neurodegeneration in the hippocampus of the adult rat. Brain Res. Bull 2005, 65, 471–477. [Google Scholar]
- Corvino, V.; Marchese, E.; Giannetti, S.; Lattanzi, W.; Bonvissuto, D.; Biamonte, F.; Mongiovì, A.M.; Michetti, F.; Geloso, M.C. The neuroprotective and neurogenic effects of neuropeptide Y administration in an animal model of hippocampal neurodegeneration and temporal lobe epilepsy induced by trimethyltin. J. Neurochem 2012, 122, 415–426. [Google Scholar]
- Harry, G.J.; Lefebvre d’Hellencourt, C. Dentate gyrus: Alterations that occur with hippocampal injury. Neurotoxicology 2003, 24, 343–356. [Google Scholar]
- Little, A.R.; Benkovic, S.A.; Miller, D.B.; O’Callaghan, J.P. Chemically induced neuronal damage and gliosis: Enhanced expression of the proinflammatory chemokine, monocyte chemoattractant protein (MCP)-1, without a corresponding increase in proinflammatory cytokines. Neuroscience 2002, 115, 307–320. [Google Scholar]
- Funk, J.A.; Gohlke, J.; Kraft, A.D.; McPherson, C.A.; Collins, J.B.; Harry, G.J. Voluntary exercise protects hippocampal neurons from trimethyltin injury: Possible role of interleukin-6 to modulate tumor necrosis factor receptor mediated neurotoxicity. Brain Behav. Immun 2011, 25, 1063–1077. [Google Scholar]
- Kraft, A.D.; Harry, G.J. Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. Int. J. Environ. Res. Public Health 2011, 8, 2980–3018. [Google Scholar]
- Koczyk, D. How does trimethyltin affect the brain: Facts and hypotheses. Acta Neurobiol. Exp. (Wars) 1996, 56, 587–596. [Google Scholar]
- Florea, A.M.; Büsselberg, D. Anti-cancer drugs interfere with intracellular calcium signaling. Neurotoxicology 2009, 30, 803–810. [Google Scholar]
- Aldrige, W.N.; Street, B.W.; Skilleter, D.N. Oxidative phosphorylation. Halide-dependent and halide-independent effects of triorganotin and triorganolead compounds on mitochondrial functions. Biochem. J 1977, 168, 353–364. [Google Scholar]
- Misiti, F.; Orsini, F.; Clementi, M.E.; Lattanzi, W.; Giardina, B.; Michetti, F. Mitochondrial oxygen consumption inhibition importance for TMT-dependent cell death in undifferentiated PC12 cells. Neurochem. Int 2008, 52, 1092–1099. [Google Scholar]
- Billingsley, M.L.; Yun, J.; Reese, B.E.; Davidson, C.E.; Buck-Koehntop, B.A.; Veglia, G. Functional and structural properties of stannin: Roles in cellular growth, selective toxicity, and mitochondrial responses to injury. J. Cell. Biochem 2006, 98, 243–250. [Google Scholar]
- Vrana, K.E.; Freeman, W.M.; Aschner, M. Use of microarray technologies in toxicology research. Neurotoxicology 2003, 24, 321–332. [Google Scholar]
- Kassed, C.A.; Butler, T.L.; Patton, G.W.; Demesquita, D.D.; Navidomskis, M.T.; Mémet, S.; Israël, A.; Pennypacker, K.R. Injury-induced NF-κB activation in the hippocampus: Implications for neuronal survival. FASEB J 2004, 18, 723–724. [Google Scholar]
- Lefebvre d’Hellencourt, C.; Harry, G.J. Molecular profiles of mRNA levels in laser capture microdissected murine hippocampal regions differentially responsive to TMT-induced cell death. J. Neurochem 2005, 93, 206–220. [Google Scholar]
- Morita, M.; Imai, H.; Liu, Y.; Xu, X.; Sadamatsu, M.; Nakagami, R.; Shirakawa, T.; Nakano, K.; Kita, Y.; Yoshida, K.; et al. FK506-protective effects against trimethyltin neurotoxicity in rats: Hippocampal expression analyses reveal the involvement of periarterial osteopontin. Neuroscience 2008, 153, 1135–1145. [Google Scholar]
- Little, A.R.; Miller, D.B.; Li, S.; Kashon, M.L.; O’Callaghan, J.P. Trimethyltin-induced neurotoxicity: Gene expression pathway analysis, q-RT-PCR and immunoblotting reveal early effects associated with hippocampal damage and gliosis. Neurotoxicol. Teratol 2012, 34, 72–82. [Google Scholar]
- Lattanzi, W.; Bernardini, C.; Gangitano, C.; Michetti, F. Hypoxia-like transcriptional activation in TMT-induced degeneration: Microarray expression analysis on PC12 cells. J. Neurochem 2007, 100, 1688–1702. [Google Scholar]
- Balaban, C.D.; O’Callaghan, J.P.; Billingsley, M.L. Trimethyltin-induced neuronal damage in the rat brain: Comparative studies using silver degeneration stains, immunocytochemistry and immunoassay for neurotypic and gliotypic proteins. Neuroscience 1988, 26, 337–361. [Google Scholar]
- Harry, G.J.; Goodrum, J.F.; Krigman, M.R.; Morell, P. The use of Synapsin I as a biochemical marker for neuronal damage by trimethyltin. Brain Res 1985, 326, 9–18. [Google Scholar]
- Moser, V.C. Rat strain-and gender-related differences in neurobehavioral screening: Acute trimethyltin neurotoxicity. J. Toxicol. Environ. Health 1996, 47, 567–586. [Google Scholar]
- Ishida, N.; Akaike, M.; Tsutsumi, S.; Kanai, A.; Masui, M.; Sadamatsu, Y.; Kuroda, Y.; Watanabe, Y.; McEwen, B.S.; Kato, N. Trimethyltin syndrome as a hippocampal degeneration model: Temporal changes and neurochemical features of seizure susceptibility and learning impairment. Neuroscience 1997, 8, 1183–1191. [Google Scholar]
- Brown, A.W.; Aldridge, W.N.; Street, B.W.; Verschoyle, R.D. The behavioural and neuropathologic sequelae of intoxication by trimethyltin compound in the rat. Am. J. Pathol 1979, 97, 59–81. [Google Scholar]
- Ishikawa, K.; Kubo, T.; Shibanoki, S.; Matsumoto, A.; Hata, H.; Asai, S. Hippocampal degeneration inducing impairment of learning in rats: Model of dementia? Behav. Brain Res 1997, 83, 39–44. [Google Scholar]
- Nilsberth, C.; Kostyszyn, B.; Luthman, J. Changes in APP, PS1 and other factors related to Alzheimer’s disease pathophysiology after trimethyltin-induced brain lesion in the rat. Neurotox. Res 2002, 4, 625–636. [Google Scholar]
- O’Connell, A.W.; Strada, O.; Earley, B.; Leonard, B.E. Altered expression of amyloid protein precursor mRNA in the rat hippocampus following trimethyltin intoxication: An in situ hybridization study. Neurochem. Int 1997, 30, 313–320. [Google Scholar]
- Woodruff, M.L.; Baisden, R.H. Trimethyltin Neurotoxicity in the Rat as an Analogous Model of Alzheimer’s Disease. In Toxin-Induced Models of Neurological Disorders; Woodruff, M.L., Nonneman, A.J., Eds.; Plenum Press: New York, NY, USA, 1994; pp. 319–335. [Google Scholar]
- Figiel, I.; Fiedorowicz, A. Trimethyltin-evoked neuronal apoptosis and glia response in mixed cultures of rat hippocampal dentate gyrus: A new model for the study of the cell type-specific influence of neurotoxins. Neurotoxicology 2002, 23, 77–86. [Google Scholar]
- Jenkins, S.M.; Barone, S. The neurotoxicant trimethyltin induces apoptosis via caspase activation, p38 protein kinase, and oxidative stress in PC12 cells. Toxicol. Lett 2004, 147, 63–72. [Google Scholar]
- Reali, C.; Scintu, F.; Pillai, R.; Donato, R.; Michetti, F.; Sogos, V. S100b counteracts effects of the neurotoxicant trimethyltin on astrocytes and microglia. J. Neurosci. Res 2005, 81, 677–686. [Google Scholar]
- Aschner, M.; Aschner, J.L. Cellular and molecular effects of trimethyltin and triethyltin: Relevance to organotin neurotoxicity. Neurosci. Biobehav. Rev 1992, 16, 427–435. [Google Scholar]
- Eskes, C.; Juillerat-Jeanneret, L.; Leuba, G.; Honegger, P.; Monnet-Tschudi, F. Involvement of microglia-neuron interactions in the tumor necrosis factor-alpha release, microglial activation, and neurodegeneration induced by trimethyltin. J. Neurosci. Res 2003, 71, 583–590. [Google Scholar]
- Figiel, I.; Dzwonek, K. TNFalpha and TNF receptor 1 expression in the mixed neuronal-glial cultures of hippocampal dentate gyrus exposed to glutamate or trimethyltin. Brain Res 2007, 1131, 17–28. [Google Scholar]
- Gunasekar, P.G.; Mickova, V.; Kotyzova, D.; Li, L.; Borowitz, J.L. Role of astrocytes in trimethyltin neurotoxicity. J. Biochem. Mol. Toxicol 2001, 15, 256–262. [Google Scholar]
- Harry, G.J.; Tyler, K.; d’Hellencourt, C.L.; Tilson, H.A.; Maier, W.E. Morphological alterations and elevations in tumor necrosis factor-alpha, interleukin (IL)-1alpha, and IL-6 in mixed glia cultures following exposure to trimethyltin: Modulation by proinflammatory cytokine recombinant proteins and neutralizing antibodies. Toxicol. Appl. Pharmacol 2002, 180, 205–218. [Google Scholar]
- Harry, G.J.; Lefebvre d’Hellencourt, C.; McPherson, C.A.; Funk, J.A.; Aoyama, M.; Wine, R.N. Tumor necrosis factor p55 and p75 receptors are involved in chemical-induced apoptosis of dentate granule neurons. J. Neurochem 2008, 106, 281–298. [Google Scholar]
- Thompson, T.A.; Lewis, J.M.; Dejneka, N.S.; Severs, W.B.; Polavarapu, R.; Billingsley, M.L. Induction of apoptosis by organotin compounds in vitro: Neuronal protection with antisense oligonucleotides directed against stannin. J. Pharmacol. Exp. Ther 1996, 276, 1201–1211. [Google Scholar]
- Ekuta, J.E.; Hikal, A.H.; Matthews, J.C. Toxicokinetics of trimethyltin in four inbred strains of mice. Toxicol. Lett 1998, 95, 41–96. [Google Scholar]
- Wenger, G.R.; McMillan, D.E.; Chang, L.W. Behavioral effects of trimethyltin in two strains of mice. II. Multiple fixed ratio, fixed interval. Toxicol. Appl. Pharmacol 1984, 73, 89–96. [Google Scholar]
- Kawada, K.; Yoneyama, M.; Nagashima, R.; Ogita, K.; Kawada, K. In vivo acute treatment with trimethyltin chloride causes neuronal degeneration in the murine olfactory bulb and anterior olfactory nucleus by different cascades in each region. J. Neurosci. Res 2008, 86, 1635–1646. [Google Scholar]
- Fiedorowicz, A.; Figiel, I.; Kaminska, B.; Zeremba, M.; Wilk, S.; Oderfeld-Nowak, B. Dentate granule neuron apoptosis and glia activation in murine hippocampus induced by trimethyltin exposure. Brain Res 2001, 912, 116–127. [Google Scholar]
- Geloso, M.C.; Vercelli, A.; Corvino, V.; Repici, M.E.; Boca, M.; Haglid, K.; Zelano, G.; Michetti, F. Cyclooxygenase-2 and caspase-3 expression in trimethyltin induced apoptosis in the mouse hippocampus. Exp. Neurol 2002, 175, 152–160. [Google Scholar]
- Ito, C.Y.; Adey, N.; Bautch, V.L.; Baldwin, A.S., Jr. Structure and evolution of the human IKBA gene. Genomics 1995, 29, 490–495. [Google Scholar]
- Kassed, C.A.; Willing, A.E.; Garbuzova-Davis, S.; Sanberg, P.R.; Pennypacker, K.R. Lack of NF-κB p50 exacerbates degeneration of hippocampal neurons after chemical exposure and impairs learning. Exp. Neurol 2002, 176, 277–288. [Google Scholar]
- Qing, Y.; Liang, Y.; Du, Q.; Fan, P.; Xu, H.; Xu, Y.; Shi, N. Apoptosis induced by Trimethyltin chloride in human neuroblastoma cells SY5Y is regulated by a balance and cross-talk between NF-κB and MAPKs signaling pathways. Arch. Toxicol 2013, 87, 1273–1285. [Google Scholar]
- Zhang, L.; Li, L.; Prabhakaran, K.; Borowitz, J.L.; Isom, G.E. Trimethyltin-induced apoptosis is associated with upregulation of inducible nitric oxide synthase and Bax in a hippocampal cell line. Toxicol. Appl. Pharmacol 2006, 216, 34–43. [Google Scholar]
- Shohami, E.; Ginis, I.; Hallenbeck, J.M. Dual role of tumor necrosis factor alpha in brain injury. Cytokine Growth Factor Rev 1999, 10, 119–130. [Google Scholar]
- Maier, W.E.; Brown, H.W.; Tilson, H.A.; Luster, M.I.; Harry, G.J. Trimethyltin increases interleukin (IL)-1α, IL-6 and tumor necrosis factor α mRNA levels in rat hippocampus. J. Neuroimmunol 1995, 59, 65–75. [Google Scholar]
- Teocchi, M.A.; Ferreira, A.É.; da Luz de Oliveira, E.P.; Tedeschi, H.; D’Souza-Li, L. Hippocampal gene expression dysregulation of Klotho, nuclear factor kappa B and tumor necrosis factor in temporal lobe epilepsy patients. J. Neuroinflammation 2013, 10. [Google Scholar] [CrossRef]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res 2005, 65, 939–947. [Google Scholar]
- Rowley, M.; van Ness, B. Activation of N-ras and K-ras induced by interleukin-6 in a myeloma cell line: Implications for disease progression and therapeutic response. Oncogene 2002, 21, 8769–8775. [Google Scholar]
- Reich, N.C. STAT dynamics. Cytokine Growth Factor Rev 2007, 18, 511–518. [Google Scholar]
- McPherson, C.A.; Kraft, A.D.; Harry, G.J. Injury-induced neurogenesis: Consideration of resident microglia as supportive of neural progenitor cells. Neurotox. Res 2011, 19, 341–352. [Google Scholar]
- Harry, G.J. Microglia during development and aging. Pharmacol. Ther 2013, 139, 313–326. [Google Scholar]
- Latini, L.; Geloso, M.C.; Corvino, V.; Giannetti, S.; Florenzano, F.; Viscomi, M.T.; Michetti, F.; Molinari, M. Trimethyltin intoxication up-regulates nitric oxide synthase in neurons and purinergic ionotropic receptor 2 in astrocytes in the hippocampus. J. Neurosci. Res 2010, 88, 500–509. [Google Scholar]
- Geloso, M.C.; Vinesi, P.; Michetti, F. Parvalbumin-immunoreactive neurons are not affected by trimethyltin-induced neurodegeneration in the rat hippocampus. Exp. Neurol 1996, 139, 269–277. [Google Scholar]
- Geloso, M.C.; Vinesi, P.; Michetti, F. Calretinin-containing neurons in trimethyltin-induced neurodegeneration in the rat hippocampus: An immunocytochemical study. Exp. Neurol 1997, 146, 67–73. [Google Scholar]
- Geloso, M.C.; Vinesi, P.; Michetti, F. Neuronal subpopulations of developing rat hippocampus containing different calcium-binding proteins behave distinctively in trimethyltin-induced neurodegeneration. Exp. Neurol 1998, 154, 645–653. [Google Scholar]
- Geloso, M.C.; Corvino, V.; Cavallo, V.; Toesca, A.; Guadagni, E.; Passalacqua, R.; Michetti, F. Expression of astrocytic nestin in the rat hippocampus during trimethyltin-induced neurodegeneration. Neurosci. Lett 2004, 357, 103–106. [Google Scholar]
- Pompili, E.; Nori, S.L.; Geloso, M.C.; Guadagni, E.; Corvino, V.; Michetti, F.; Fumagalli, L. Trimethyltin-induced differential expression of PAR subtypes in reactive astrocytes of the rat hippocampus. Brain Res. Mol. Brain Res 2004, 122, 93–98. [Google Scholar]
- Brabeck, C.; Michetti, F.; Geloso, M.C.; Corvino, V.; Goezalan, F.; Meyermann, R.; Schluesener, H.J. Expression of EMAP-II by activated monocytes/microglial cells in different regions of the rat hippocampus after trimethyltin-induced brain damage. Exp. Neurol 2002, 177, 341–346. [Google Scholar]
- Pompili, E.; Fabrizi, C.; Nori, S.L.; Panetta, B.; Geloso, M.C.; Corvino, V.; Michetti, F.; Fumagalli, L. Protease-activated receptor-1 expression in rat microglia after trimethyltin treatment. J. Histochem. Cytochem 2011, 59, 302–311. [Google Scholar]
- Brock, T.O.; O’Callaghan, J.P. Quantitative changes in the synaptic vesicle proteins synapsin I and p38 and the astrocyte-specific protein glial fibrillary acidic protein are associated with chemical-induced injury to the rat central nervous system. J. Neurosci 1987, 7, 931–942. [Google Scholar]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar]
- Steinacker, P.; Aitken, A.; Otto, M. 14-3-3 proteins in neurodegeneration. Semin. Cell Dev. Biol 2011, 22, 696–704. [Google Scholar]
- Yamashima, T. Reconsider Alzheimer’s disease by the ‘calpain-cathepsin hypothesis’— A perspective review. Prog. Neurobiol 2013, 105, 1–23. [Google Scholar]
- Wang, B.; Ling, S.; Lin, W.C. 14-3-3T regulates Beclin 1 and is required for autophagy. PLoS One 2010, 5, e10409. [Google Scholar]
- Fabrizi, C.; Somma, F.; Pompili, E.; Biagioni, F.; Lenzi, P.; Fornai, F.; Fumagalli, L. Role of autophagy inhibitors and inducers in modulating the toxicity of trimethyltin in neuronal cell cultures. J. Neural Transm 2012, 119, 1295–1305. [Google Scholar]
- Viscomi, M.T.; D’Amelio, M. The “Janus-faced role” of autophagy in neuronal sickness: Focus on neurodegeneration. Mol. Neurobiol 2012, 46, 513–521. [Google Scholar]
- Stoorvogel, W.; Kleijmeer, M.J.; Geuze, H.J.; Raposo, G. The biogenesis and functions of exosomes. Traffic 2002, 3, 321–330. [Google Scholar]
- McCue, H.V.; Wardyn, J.D.; Burgoyne, R.D.; Haynes, L.P. Source generation and characterization of a lysosomally targeted, genetically encoded Ca2+-sensor. Biochem. J 2013, 49, 449–457. [Google Scholar]
- Kipanyula, M.J.; Contreras, L.; Zampese, E.; Lazzari, C.; Wong, A.K.; Pizzo, P.; Fasolato, C.; Pozzan, T. Ca2+ dysregulation in neurons from transgenic mice expressing mutant presenilin 2. Aging Cell 2012, 11, 885–893. [Google Scholar]
- Florea, A.; Elke, D.; Busselberg, D. Elevated Ca2+ transients induced by trimethyltin chloride in HeLa cells: Types and levels of response. Cell Calcium 2005, 37, 251–258. [Google Scholar]
- Florea, A.M.; Splettstoesser, F.; Dopp, E.; Rettenmeier, A.W.; Busselberg, D. Modulation of intracellular calcium homeostasis by trimethyltin chloride in human tumour cells: Neuroblastoma SY5Y and cervix adenocarcinoma HeLaS3. Toxicology 2005, 216, 1–8. [Google Scholar]
- Piacentini, R.; Gangitano, C.; Ceccariglia, S.; Del Fà, A.; Azzena, G.B.; Michetti, F.; Grassi, C. Dysregulation of intracellular calcium homeostasis is responsible for neuronal death in an experimental model of selective hippocampal degeneration induced by trimethyltin. J. Neurochem 2008, 105, 2109–2121. [Google Scholar]
- Ceccariglia, S.; D’Altocolle, A.; Del Fà, A.; Pizzolante, F.; Caccia, E.; Michetti, F.; Gangitano, C. Cathepsin D plays a crucial role in the trimethyltin-induced hippocampal neurodegeneration process. Neuroscience 2011, 174, 160–170. [Google Scholar]
- Shibasaki, F.; Hallin, U.; Uchino, H. Calcineurin as a multifunctional regulator. J. Biochem 2002, 131, 1–15. [Google Scholar]
- Usui, T.; Shima, Y.; Shimada, Y.; Hirano, S.; Burgess, R.W.; Schwarz, T.L.; Takeichi, M.; Uemura, T. Flamingo, a seven-pass transmembrane cadherin, regulates planar cell polarity under the control of Frizzled. Cell 1999, 98, 585–595. [Google Scholar]
- Pennypacker, K.R.; Kassed, C.A.; Eidizadeh, S.; Saporta, S.; Sanberg, P.R.; Willing, A.E. NF-κB p50 is increased in neurons surviving hippocampal injury. Exp. Neurol 2001, 172, 307–319. [Google Scholar]
- Denhardt, D.T.; Noda, M.; O’Regan, A.W.; Pavlin, D.; Berman, J.S. Osteopontin as a means to cope with environmental insults: Regulation of inflammation, tissue remodeling, and cell survival. J. Clin. Invest 2001, 107, 1055–1061. [Google Scholar]
- Borges, K.; Gearing, M.; Rittling, S.; Sorensen, E.S.; Kotloski, R.; Denhardt, D.T.; Dingledine, R. Characterization of osteopontin expression and function after status epilepticus. Epilepsia 2008, 49, 1675–1685. [Google Scholar]
- Jahnke, G.D.; Brunssen, S.; Maier, W.E.; Harry, G.J. Neurotoxicant-induced elevation of adrenomedullin expression in hippocampus and glia cultures. J. Neurosci. Res 2001, 66, 464–474. [Google Scholar]
- Harry, G.J.; Sills, R.; Schlosser, M.J.; Maier, W.E. Neurodegeneration and glia response in rat hippocampus following nitro-l-arginine methyl ester (L-NAME). Neurotox. Res 2001, 3, 307–319. [Google Scholar]
- Liu, Y.; Imai, H.; Sadamatsu, M.; Tsunashima, K.; Kato, N. Cytokines participate in neuronal death induced by trimethyltin in the rat hippocampus via type II glucocorticoid receptors. Neurosci. Res 2005, 51, 319–327. [Google Scholar]
- Corvino, V.; Marchese, E.; Zarkovic, N.; Zarkovic, K.; Cindric, M.; Waeg, G.; Michetti, F.; Geloso, M.C. Distribution and timecourse of 4-hydroxynonenal, heat shock protein 110/105 family members and cyclooxygenase-2 expression in the hippocampus of rat during trimethyltin-induced neurodegeneration. Neurochem. Res 2011, 36, 1490–1500. [Google Scholar]
- Bruccoleri, A.; Brown, H.; Harry, G.J. Cellular localization and temporal elevation of tumor necrosis factor-alpha, interleukin-1 alpha, and transforming growth factor-beta 1 mRNA in hippocampal injury response induced by trimethyltin. J. Neurochem 1998, 71, 1577–1587. [Google Scholar]
- Zhuang, S. Regulation of STAT signaling by acetylation. Cell. Signal 2013, 25, 1924–1931. [Google Scholar]
- Wine, R.N.; McPherson, C.A.; Harry, G.J. IGF-1 and pAKT signaling promote hippocampal CA1 neuronal survival following injury to dentate granule cells. Neurotox. Res 2009, 16, 280–292. [Google Scholar]
- Suh, H.S.; Zhao, M.L.; Derico, L.; Choi, N.; Lee, S.C. Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in human microglia: Differential regulation by inflammatory mediators. J. Neuroinflammation 2013, 10. [Google Scholar] [CrossRef]
- Rizo, J.; Chen, X.; Arac, D. Unraveling themechanisms of synaptotagmin and SNARE function in neurotransmitter release. Trends Cell Biol 2006, 16, 339–350. [Google Scholar]
- Ishizuka, T.; Saisu, H.; Odani, S.; Abe, T. Synaphin: A protein associated with the docking/fusion complex in presynaptic terminal. Biochem. Biophys. Res. Commun 1995, 213, 1107–1114. [Google Scholar]
- Baumgartner, W.; Osmanagic, A.; Gebhard, M.; Kraemer, S.; Golenhofen, N. Different pH-dependencies of the two synaptic adhesion molecules N-cadherin and cadherin-11 and the possible functional implication for long term potentiation. Synapse 2013. [Google Scholar] [CrossRef]
- Guimbal, C.; Klostermann, A.; Kilimann, M.W. Phylogenetic conservation of 4-aminobutyric acid [GABA] transporter isoforms. Cloning and pharmacological characterization of a GABA/β-alanine transporter from Torpedo. Eur. J. Biochem 1995, 234, 794–800. [Google Scholar]
- Singh, N.; Sharma, G.; Mishra, V. Hypoxia inducible factor-1: Its potential role in cerebral ischemia. Cell. Mol. Neurobiol 2012, 32, 491–507. [Google Scholar]
- Cunningham, L.A.; Candelario, K.; Li, L. Roles for HIF-1α in neural stem cell function and the regenerative response to stroke. Behav. Brain Res 2012, 227, 410–417. [Google Scholar]
- Harkins, A.B.; Armstrong, D.L. Trimethyltin alters membrane properties of CA1 hippocampal neurons. Neurotoxicology 1992, 13, 569–581. [Google Scholar]
- Chow, S.C.; Orrenius, S. Rapid cytoskeleton modification in thymocytes induced by the immunotoxicant tributyltin. Toxicol. Appl. Pharmacol 1994, 127, 19–26. [Google Scholar]
- Richter-Landsberg, C.; Besser, A. Effects of organotins on rat brain astrocytes in culture. J. Neurochem 1994, 63, 2202–2209. [Google Scholar]
- Ortiz, A.; Teruel, J.A.; Aranda, F.J. Effect of triorganotin compounds on membrane permeability. Biochim. Biophys. Acta 2005, 1720, 137–142. [Google Scholar]
- Bernardini, C.; Barba, M.; Tamburrini, G.; Massimi, L.; di Rocco, C.; Michetti, F.; Lattanzi, W. Gene expression profiling in human craniosynostoses: A tool to investigate the molecular basis of suture ossification. Childs Nerv. Syst 2012, 28, 1295–1300. [Google Scholar]
- Bernardini, C.; Censi, F.; Lattanzi, W.; Barba, M.; Calcagnini, G.; Giuliani, A.; Tasca, G.; Sabatelli, M.; Ricci, E.; Michetti, F. Mitochondrial network genes in the skeletal muscle of amyotrophic lateral sclerosis patients. PLoS One 2013, 8, e57739. [Google Scholar]
- Bernardini, C.; Lattanzi, W.; Bosco, P.; Franceschini, C.; Plazzi, G.; Michetti, F.; Ferri, R. Genome-wide gene expression profiling of human narcolepsy. Gene Expr 2012, 15, 171–181. [Google Scholar]
- Bernardini, C.; Lattanzi, W.; Businaro, R.; Leone, S.; Corvino, V.; Sorci, G.; Lauro, G.; Fumagalli, L.; Donato, F.R.; Michetti, F. Transcritpional effects of S100B on neuroblastoma cells: Perturbation of cholesterol homeostasis and interference on the cell cycle. Gene Expr 2010, 14, 345–359. [Google Scholar]
- Xiao, G. Autophagy and NF-κB: Fight for fate. Cytokine Growth Factor Rev 2007, 18, 233–243. [Google Scholar]



{kind=link}
{kind=link}
| Model animal (No. of samples per experimental group) | Age at treatment | Rodent strain – Animal gender a | TMT dosage (administration route) | Hippocampal tissue specimen | Tested time points (post-TMT treatment) | Reference |
|---|---|---|---|---|---|---|
| Mouse (n = 3) | Adult | B6, 129 Nfkb1 tmlBal B6,129 2/J – F/M | p-50 null: 2.0 mg/kg; Non transgenic: 2.25 mg/kg | Whole hippocampus | 7 days | [23] |
| Mouse (n = 10) | P21 | CD-1 – M | 3 mg/Kg (i.p.) | Microdissected DG and CA | 6–18 h | [24] |
| Mouse (n = 3) | P120 | CD-1 – M | 2.4 mg/Kg (i.p.) | Whole hippocampus | 24 h | [15] |
| Mouse (n = 3) | P21 1 year | CD-1 – M | 2.3 mg/Kg (i.p.) | Microdissected SGZ | 48 h | [10] |
| Rat (n = 3) | 6 weeks | Sprague–Dawley – M | 9 mg/Kg (oral) | Whole hippocampus | 2–5 days | [25] |
| Rat (n = 3) | 6 weeks | Long –Evans – F/M | 8.0 mg/Kg (i.p.) | Whole hippocampus | 3–5 days | [26] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lattanzi, W.; Corvino, V.; Di Maria, V.; Michetti, F.; Geloso, M.C. Gene Expression Profiling as a Tool to Investigate the Molecular Machinery Activated during Hippocampal Neurodegeneration Induced by Trimethyltin (TMT) Administration. Int. J. Mol. Sci. 2013, 14, 16817-16835. https://doi.org/10.3390/ijms140816817
Lattanzi W, Corvino V, Di Maria V, Michetti F, Geloso MC. Gene Expression Profiling as a Tool to Investigate the Molecular Machinery Activated during Hippocampal Neurodegeneration Induced by Trimethyltin (TMT) Administration. International Journal of Molecular Sciences. 2013; 14(8):16817-16835. https://doi.org/10.3390/ijms140816817
Chicago/Turabian StyleLattanzi, Wanda, Valentina Corvino, Valentina Di Maria, Fabrizio Michetti, and Maria Concetta Geloso. 2013. "Gene Expression Profiling as a Tool to Investigate the Molecular Machinery Activated during Hippocampal Neurodegeneration Induced by Trimethyltin (TMT) Administration" International Journal of Molecular Sciences 14, no. 8: 16817-16835. https://doi.org/10.3390/ijms140816817
APA StyleLattanzi, W., Corvino, V., Di Maria, V., Michetti, F., & Geloso, M. C. (2013). Gene Expression Profiling as a Tool to Investigate the Molecular Machinery Activated during Hippocampal Neurodegeneration Induced by Trimethyltin (TMT) Administration. International Journal of Molecular Sciences, 14(8), 16817-16835. https://doi.org/10.3390/ijms140816817