Trastuzumab-Peptide Interactions: Mechanism and Application in Structure-Based Ligand Design

Abstract
:1. Introduction
2. Results and Discussion
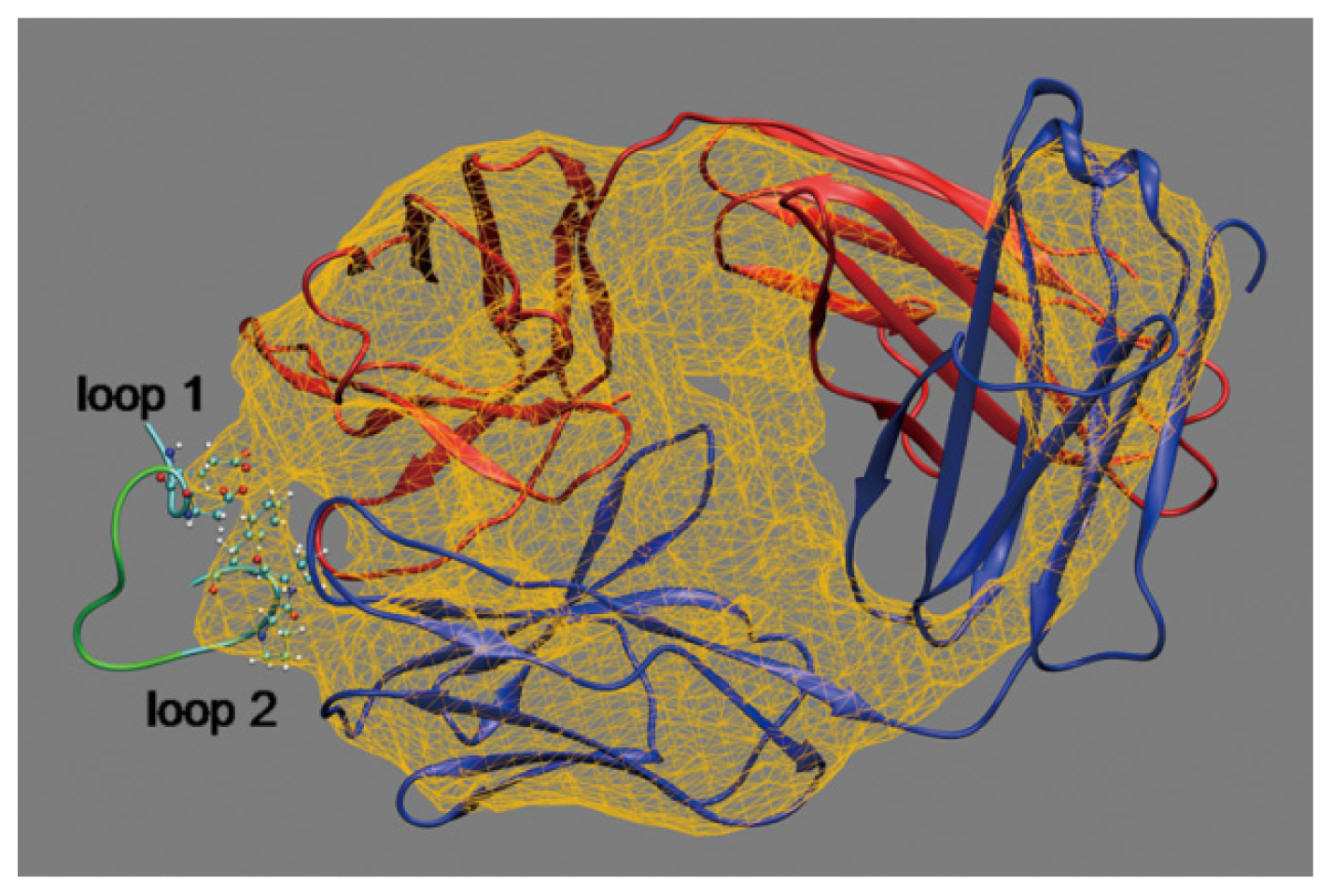
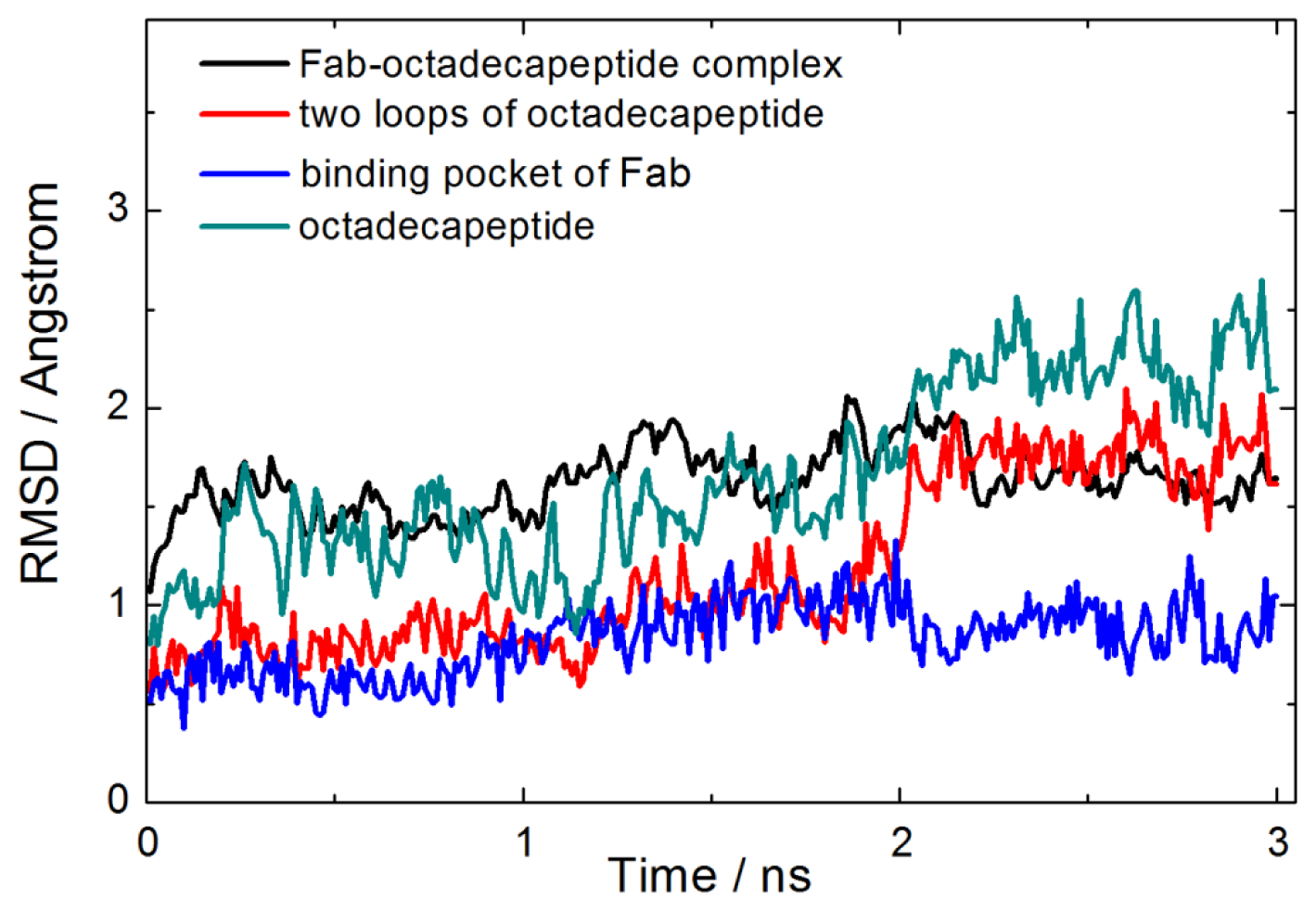
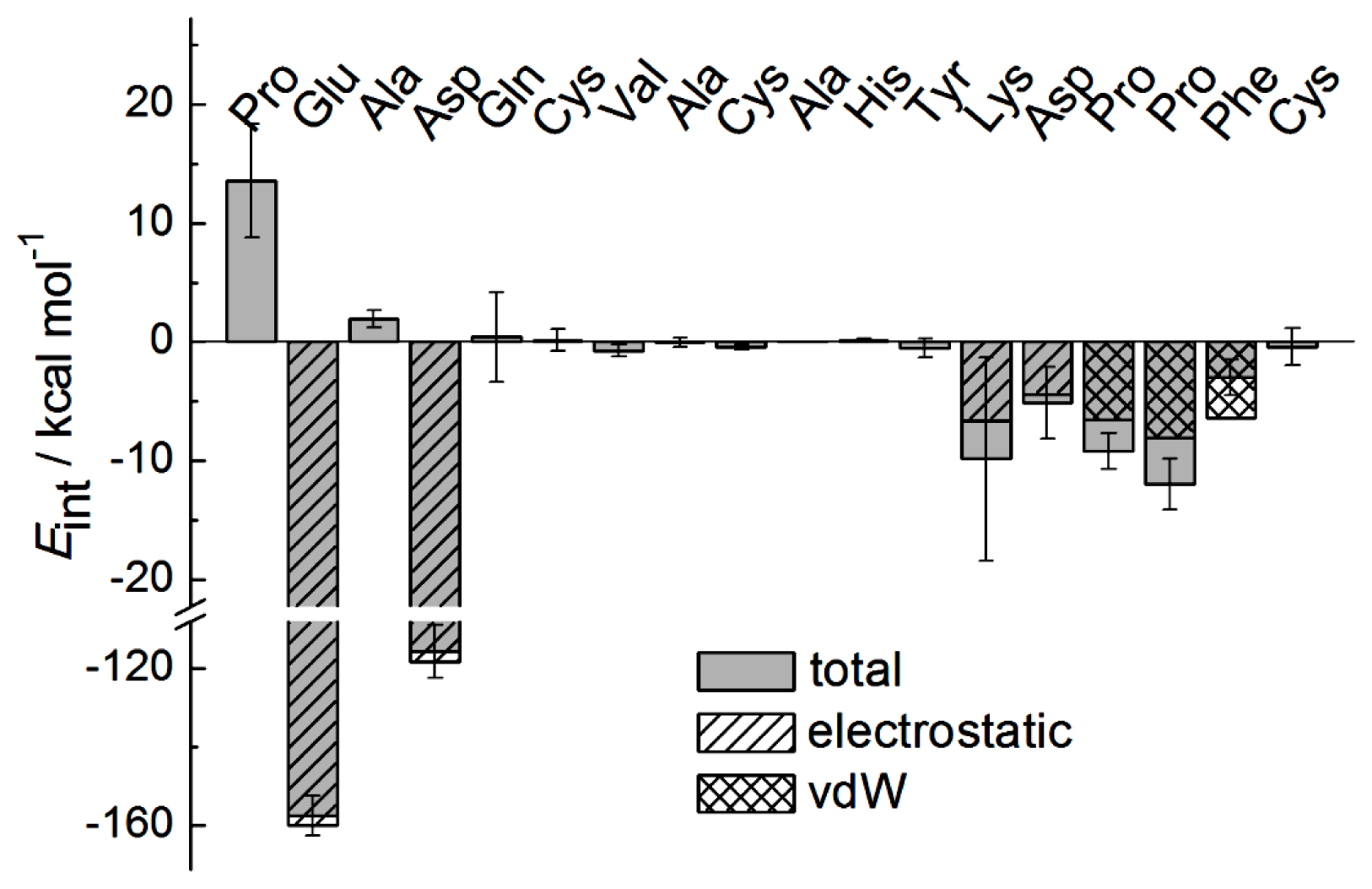
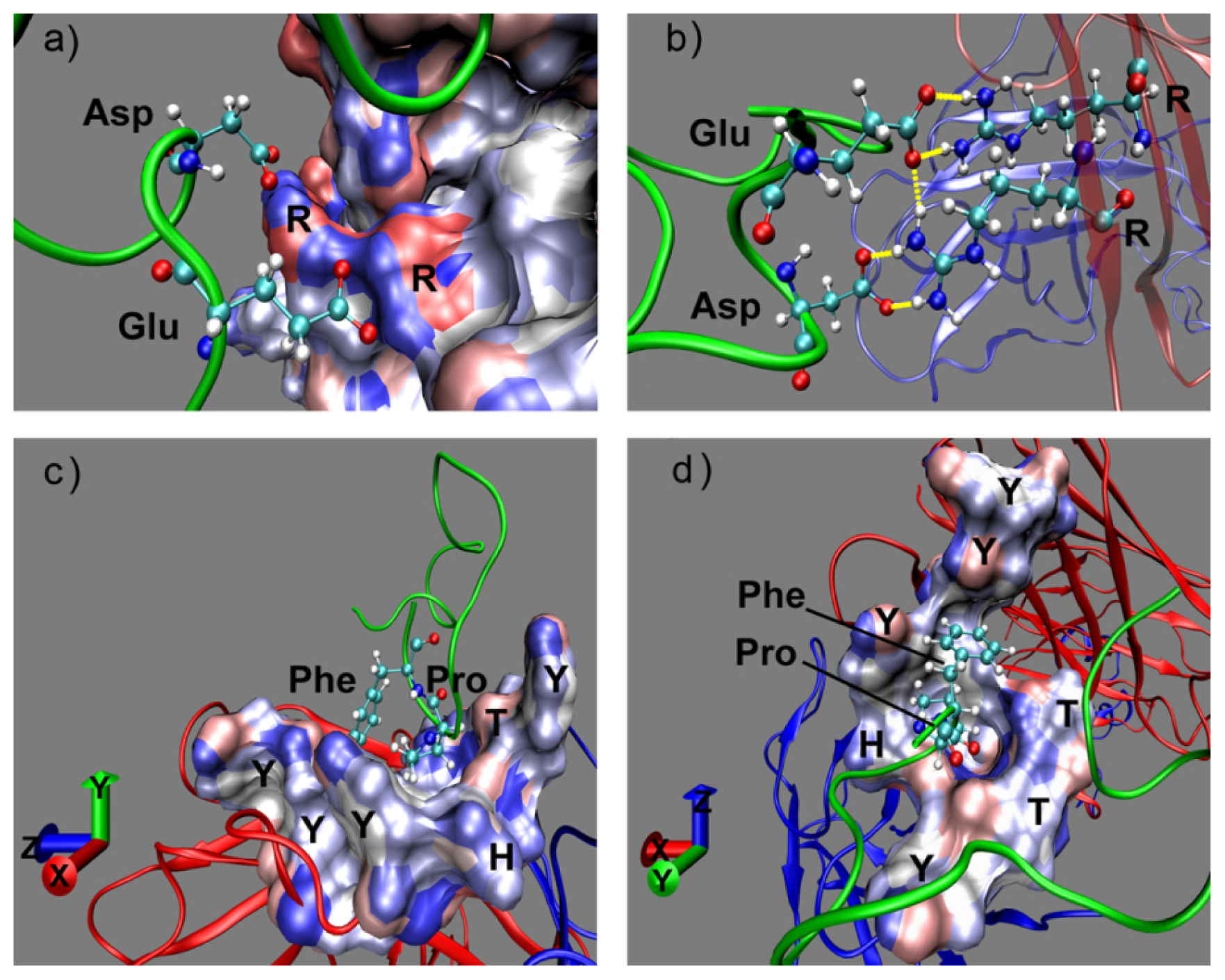
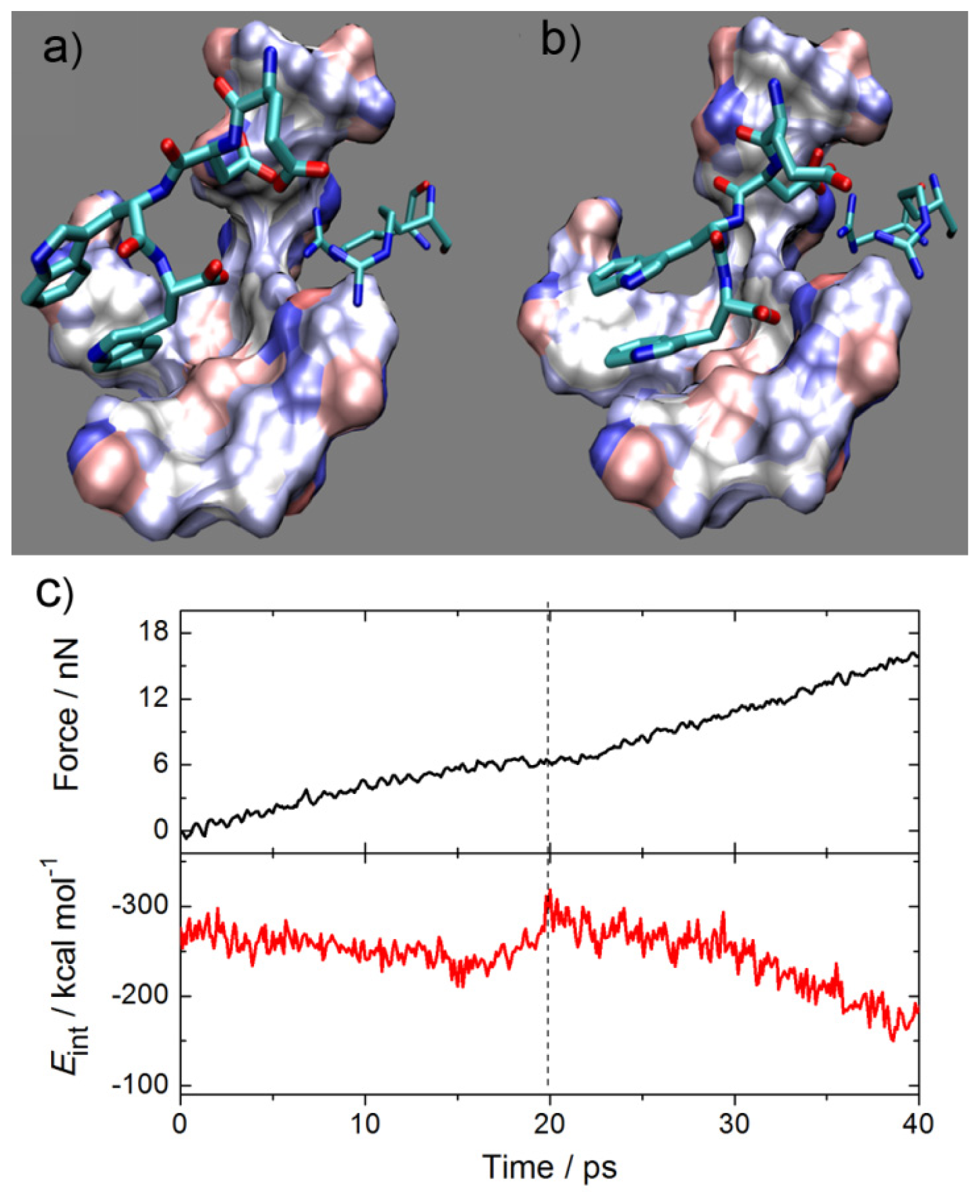
2.1. Interactions between Fab and Octadecapeptide Ligand
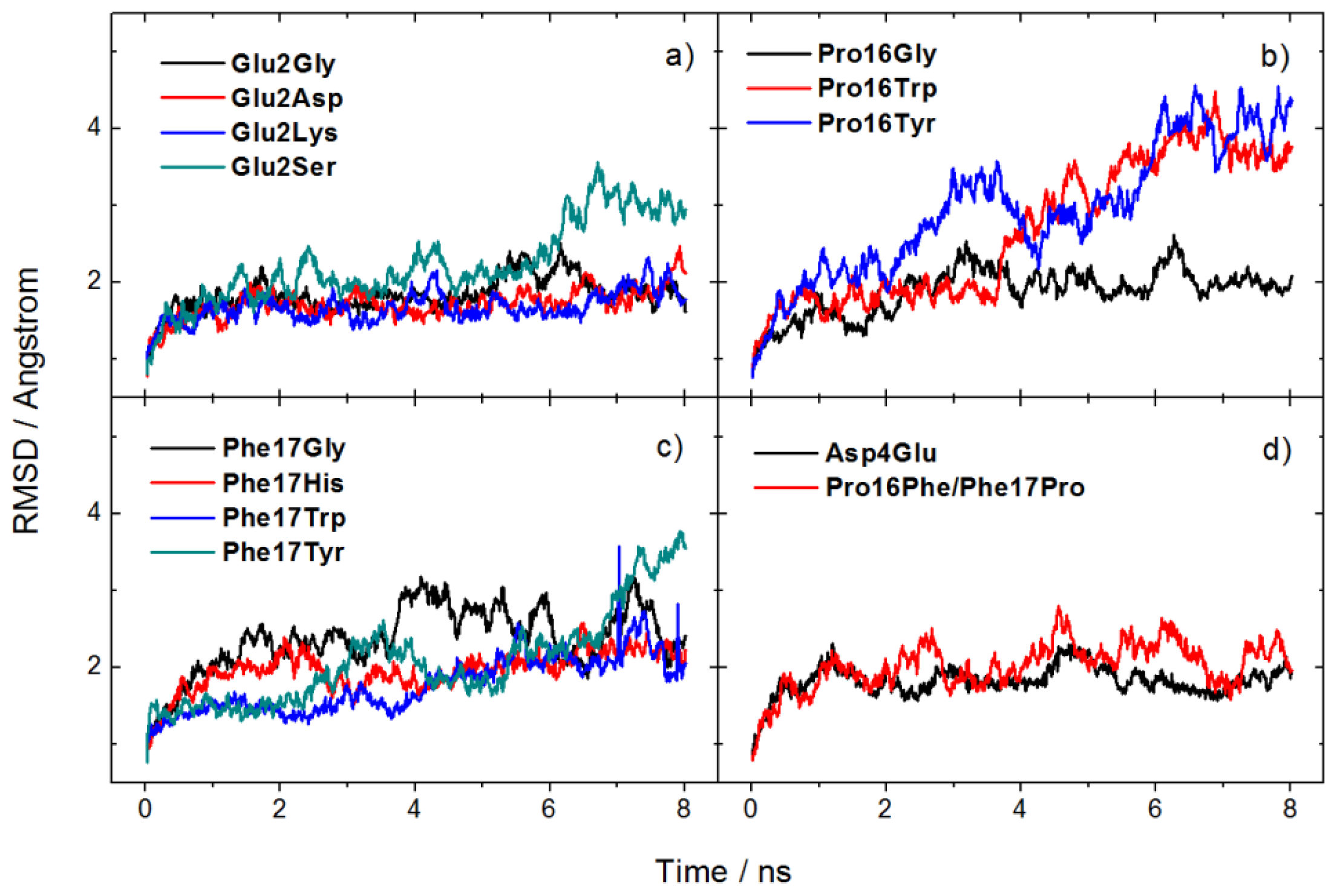
2.2. Mutation
2.3. Design and Evaluation of Tetrapeptide Ligands
3. Experimental Section
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Keskin, O.; Ma, B.; Nussinov, R. Hot regions in protein–protein interactions: The organization and contribution of structurally conserved hot spot residues. J. Mol. Biol 2005, 345, 1281–1294. [Google Scholar]
- Choi, E.J.; Mao, J.; Mayo, S.L. Computational design and biochemical characterization of maize nonspecific lipid transfer protein variants for biosensor applications. Protein Sci 2007, 16, 582–588. [Google Scholar]
- Hall, B.; Hesselberth, J.R.; Ellington, A.D. Computational selection of nucleic acid biosensors via a slip structure model. Biosens. Bioelectron 2007, 22, 1939–1947. [Google Scholar]
- Michelow, I.C.; Dong, M.; Mungall, B.A.; Yantosca, L.M.; Lear, C.; Ji, X.; Karpel, M.; Rootes, C.L.; Brudner, M.; Houen, G.; et al. A novel L-ficolin/mannose-binding lectin chimeric molecule with enhanced activity against ebola virus. J. Biol. Chem 2010, 285, 24729–24739. [Google Scholar]
- Fields, S.; Song, O. A novel genetic system to detect protein-protein interactions. Nature 1989, 340, 245–246. [Google Scholar]
- Uetz, P.; Giot, L.; Cagney, G.; Mansfield, T.A.; Judson, R.S.; Knight, J.R.; Lockshon, D.; Narayan, V.; Srinivasan, M.; Pochart, P.; et al. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature 2000, 403, 623–627. [Google Scholar]
- DeArmond, P.D.; Xu, Y.; Strickland, E.C.; Daniels, K.G.; Fitzgerald, M.C. Thermodynamic analysis o protein–ligand interactions in complex biological mixtures using a shotgun proteomics approach. J. Proteome. Res 2011, 10, 4948–4958. [Google Scholar]
- Baselga, J.; Norton, L.; Albanell, J.; Kim, Y.M.; Mendelsohn, J. Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res 1998, 58, 2825–2831. [Google Scholar]
- Dillman, R.O. Perceptions of Herceptin®: A monoclonal antibody for the treatment of breast cancer. Cancer Biother. Radiopharm 1999, 14, 5–10. [Google Scholar]
- Van Cutsem, E.; Kang, Y.; Chung, H.; Shen, L.; Sawaki, A.; Lordick, F.; Hill, J.; Lehle, M.; Feyereislova, A.; Bang, Y. Efficacy results from the ToGA trial: A phase III study of trastuzumab added to standard chemotherapy (CT) in first-line human epidermal growth factor receptor 2 (HER2)-positive advanced gastric cancer (GC). J. Clin. Oncol. 2009, 27. abstract LBA4509. [Google Scholar]
- Hage, D.S. Handbook of Affinity Chromatography, 2nd ed; Taylor & Francis: Boca Raton, FL, USA, 2006; p. 944. [Google Scholar]
- Roque, A.C.A.; Silva, C.S.O.; Taipa, M.A. Affinity-based methodologies and ligands for antibody purification: Advances and perspectives. J. Chromatogr. A 2007, 1160, 44–55. [Google Scholar]
- Scott, J.K.; Smith, G.P. Searching for peptide ligands with an epitope library. Science 1990, 249, 386–390. [Google Scholar]
- Cwirla, S.E.; Peters, E.A.; Barrett, R.W.; Dower, W.J. Peptides on phage: A vast library of peptides for identifying ligands. Proc. Natl. Acad. Sci. USA 1990, 87, 6378–6382. [Google Scholar]
- Devlin, J.J.; Panganiban, L.C.; Devlin, P.E. Random peptide libraries: A source of specific protein binding molecules. Science 1990, 249, 404–406. [Google Scholar]
- Lam, K.S.; Salmon, S.E.; Hersh, E.M.; Hruby, V.J.; Kazmierski, W.M.; Knapp, R.J. A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. [Google Scholar]
- Lam, K.S.; Lebl, M.; Krchnak, V. The “one-bead-one-compound” combinatorial library method. Chem. Rev 1997, 97, 411–448. [Google Scholar]
- Yang, H.; Gurgel, P.V.; Carbonell, R.G. Hexamer peptide affinity resins that bind the Fc region of human immunoglobulin G. J. Pept. Res 2005, 66, 120–137. [Google Scholar]
- Yang, H.O.; Gurgel, P.V.; Carbonell, R.G. Purification of human immunoglobulin G via Fc-specific small peptide ligand affinity chromatography. J. Chromatogr. A 2009, 1216, 910–918. [Google Scholar]
- Szardenings, M. Phage display of random peptide libraries: Applications, limits, and potential. J. Recept. Signal Transduct 2003, 23, 307–349. [Google Scholar]
- Deng, N.J.; Zhang, P.; Cieplak, P.; Lai, L. Elucidating the energetics of entropically driven protein-ligand association: calculations of absolute binding free energy and entropy. J. Phys. Chem. B 2011, 115, 11902–11910. [Google Scholar]
- Zhan, J.-H.; Zhao, X.I.; Huang, X.-R.; Sun, C.-C. Molecular dynamics and free energy analyses of ERK2-pyrazolylpyrrole inhibitors interactions: Insight into structure-based ligand design. J. Theor. Comput. Chem 2009, 8, 887–908. [Google Scholar]
- Singh, N.; Warshel, A. Absolute binding free energy calculations: On the accuracy of computational scoring of protein–ligand interactions. Proteins 2010, 78, 1705–1723. [Google Scholar]
- Schreier, B.; Stumpp, C.; Wiesner, S.; Hocker, B. Computational design of ligand binding is not a solved problem. Proc. Natl. Acad. Sci. USA 2009, 106, 18491–18496. [Google Scholar]
- Muegge, I.; Rarey, M. Small Molecule Docking and Scoring. In Reviews in Computational Chemistry; John Wiley & Sons: New York, NY, USA, 2001; Volume 17. [Google Scholar]
- Freed, A.S.; Garde, S.; Cramer, S.M. Molecular simulations of multimodal ligand-protein binding: Elucidation of binding sites and correlation with experiments. J. Phys. Chem. B 2011, 115, 13320–13327. [Google Scholar]
- Chen, X.; Wu, T.; Wang, Q.; Shen, J.W. Shield effect of silicate on adsorption of proteins onto silicon-doped hydroxyapatite (100) surface. Biomaterials 2008, 29, 2423–2432. [Google Scholar]
- Shen, J.W.; Wu, T.; Wang, Q.; Pan, H.H. Molecular simulation of protein adsorption and desorption on hydroxyapatite surfaces. Biomaterials 2008, 29, 513–532. [Google Scholar]
- Cho, H.S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney, D.W.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760. [Google Scholar]
- Satyanarayanajois, S.; Villalba, S.; Jianchao, L.; Lin, G.M. Design, synthesis, and docking studies of peptidomimetics based on HER2-Herceptin binding site with potential antiproliferative activity against breast cancer cell lines. Chem. Biol. Drug Des 2009, 74, 246–257. [Google Scholar]
- Latypov, R.F.; Liu, D.J.; Gunasekaran, K.; Harvey, T.S.; Razinkov, V.I.; Raibekas, A.A. Structural and thermodynamic effects of ANS binding to human interleukin-1 receptor antagonist. Protein Sci 2008, 17, 652–663. [Google Scholar]
- Celej, M.S.; Montich, C.G.; Fidelio, G.D. Protein stability induced by ligand binding correlates with changes in protein flexibility. Protein Sci 2003, 12, 1496–1506. [Google Scholar]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Hot spots—A review of the protein-protein interface determinant amino-acid residues. Proteins 2007, 68, 803–812. [Google Scholar]
- Bahadur, R.P.; Chakrabarti, P.; Rodier, F.; Janin, J. A dissection of specific and non-specific protein-protein interfaces. J. Mol. Biol 2004, 336, 943–955. [Google Scholar]
- Lund, L.N.; Gustavsson, P.E.; Michael, R.; Lindgren, J.; Norskov-Lauritsen, L.; Lund, M.; Houen, G.; Staby, A.; St Hilaire, P.M. Novel peptide ligand with high binding capacity for antibody purification. J. Chromatogr. A 2012, 1225, 158–167. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [Google Scholar]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar]
- Hirschfelder, J.O.; Curtiss, C.F.; Bird, R.B. Molecular theory of gases and liquids. Phys. Today 1955, 8, 17. [Google Scholar]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys 1995, 103, 4613–4621. [Google Scholar]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys 1994, 101, 4177–4189. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- Zhang, F.; Peng, Q.S.; Hu, M.; Wu, T. Calculation of protein molecular field based on quantum chemistry. J. Comput. Aided Des. Comput. Graph 2008, 20, 1238–1245. [Google Scholar]
- Scott, A.P.; Radom, L. Harmonic vibrational frequencies: An evaluation of Hartree–Fock, Moller–Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors. J. Phys. Chem 1996, 100, 16502–16513. [Google Scholar]
- Kang, Y.; Liu, Y.C.; Wang, Q.; Shen, J.W.; Wu, T.; Guan, W.J. On the spontaneous encapsulation of proteins in carbon nanotubes. Biomaterials 2009, 30, 2807–2815. [Google Scholar]
- Suite 2012: Prime, Version 3.1; Schrödinger, LLC: New York, NY, USA, 2012.
- Lyne, P.D.; Lamb, M.L.; Saeh, J.C. Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J. Med. Chem 2006, 49, 4805–4808. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Interaction energy (kcal/mol) | Hydrogen bond † |
|---|---|---|
| origin | −298.4 ± 12.7 | 3 |
| Glu2Gly | −224.7 ± 18.0 | 1 |
| Glu2Asp | −300.1 ± 21.0 | 2 |
| Glu2Lys | −117.2 ± 17.8 | 0 |
| Glu2Ser | −178.6 ± 20.4 | 1 |
| Asp4Glu | −268.4 ± 16.6 | 2.5 |
| Pro16Gly | −281.0 ± 23.2 | 3 |
| Pro16Trp | −302.6 ± 16.1 | 2.5 |
| Pro16Tyr | −254.7 ± 16.8 | 2.5 |
| Phe17Gly | −310.7 ± 23.5 | 2.5 |
| Phe17His | −294.6 ± 16.2 | 2.5 |
| Phe17Trp | −396.6 ± 19.8 | 3 |
| Phe17Tyr | −177.6 ± 16.9 | 3 |
| Pro16Phe | −315.2 ± 18.0 | 3 |
| Phe17Pro |
| Ligand | EDGW | EDPW | EDWW |
|---|---|---|---|
| Eint | −262.0 ± 19.0 | −295.1 ± 22.1 | −190.1 ± 17.0 |
| ΔGbind | −49.0 ± 4.8 | −73.0 ± 6.2 | −49.6 ± 5.3 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sun, T.-Y.; Wang, Q.; Zhang, J.; Wu, T.; Zhang, F. Trastuzumab-Peptide Interactions: Mechanism and Application in Structure-Based Ligand Design. Int. J. Mol. Sci. 2013, 14, 16836-16850. https://doi.org/10.3390/ijms140816836
Sun T-Y, Wang Q, Zhang J, Wu T, Zhang F. Trastuzumab-Peptide Interactions: Mechanism and Application in Structure-Based Ligand Design. International Journal of Molecular Sciences. 2013; 14(8):16836-16850. https://doi.org/10.3390/ijms140816836
Chicago/Turabian StyleSun, Tian-Yang, Qi Wang, Jin Zhang, Tao Wu, and Fan Zhang. 2013. "Trastuzumab-Peptide Interactions: Mechanism and Application in Structure-Based Ligand Design" International Journal of Molecular Sciences 14, no. 8: 16836-16850. https://doi.org/10.3390/ijms140816836
APA StyleSun, T. -Y., Wang, Q., Zhang, J., Wu, T., & Zhang, F. (2013). Trastuzumab-Peptide Interactions: Mechanism and Application in Structure-Based Ligand Design. International Journal of Molecular Sciences, 14(8), 16836-16850. https://doi.org/10.3390/ijms140816836