Bioinformatic Prediction of Gene Functions Regulated by Quorum Sensing in the Bioleaching Bacterium Acidithiobacillus ferrooxidans



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
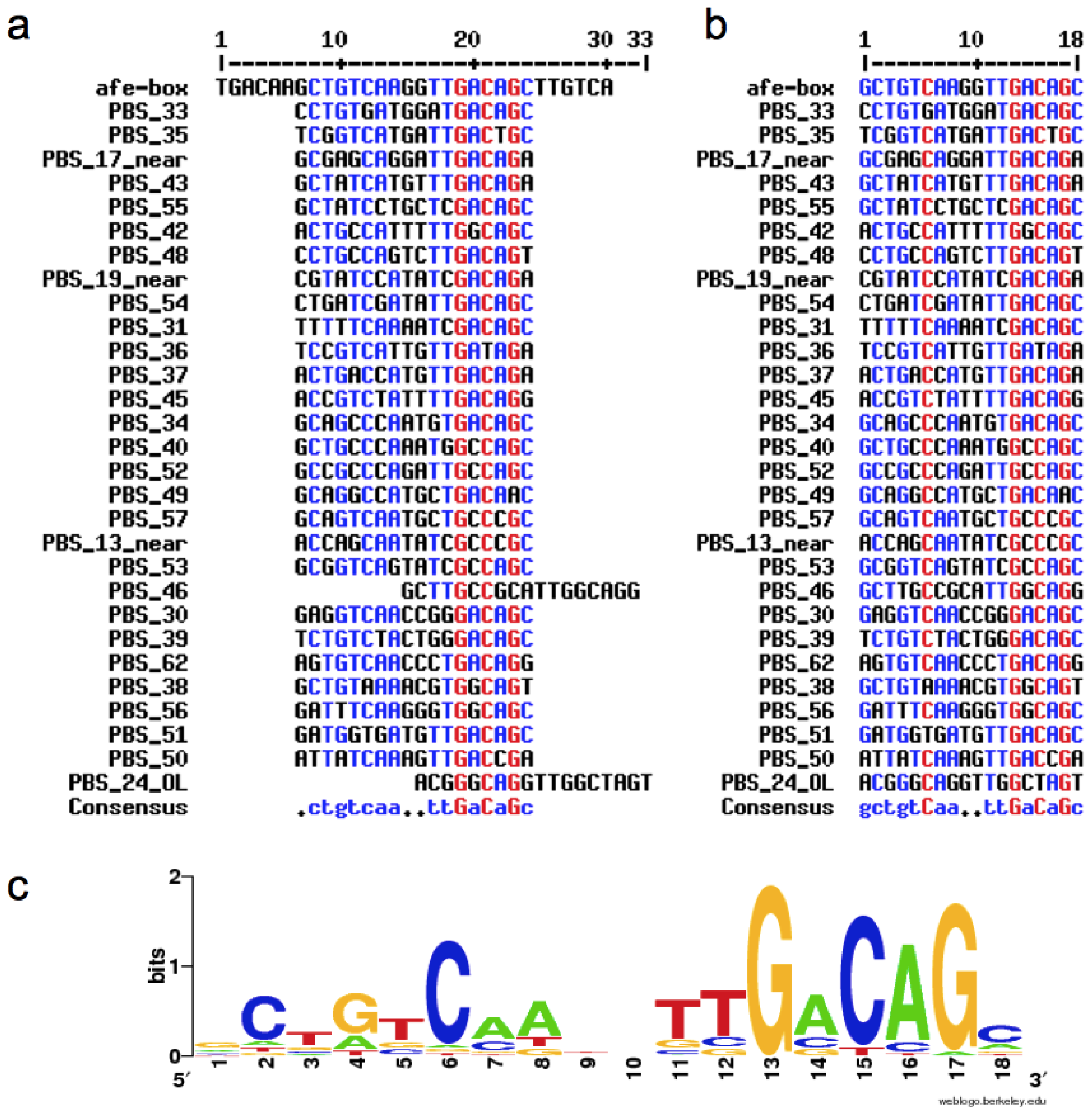
2.1. Construction of the afe-box HMMs
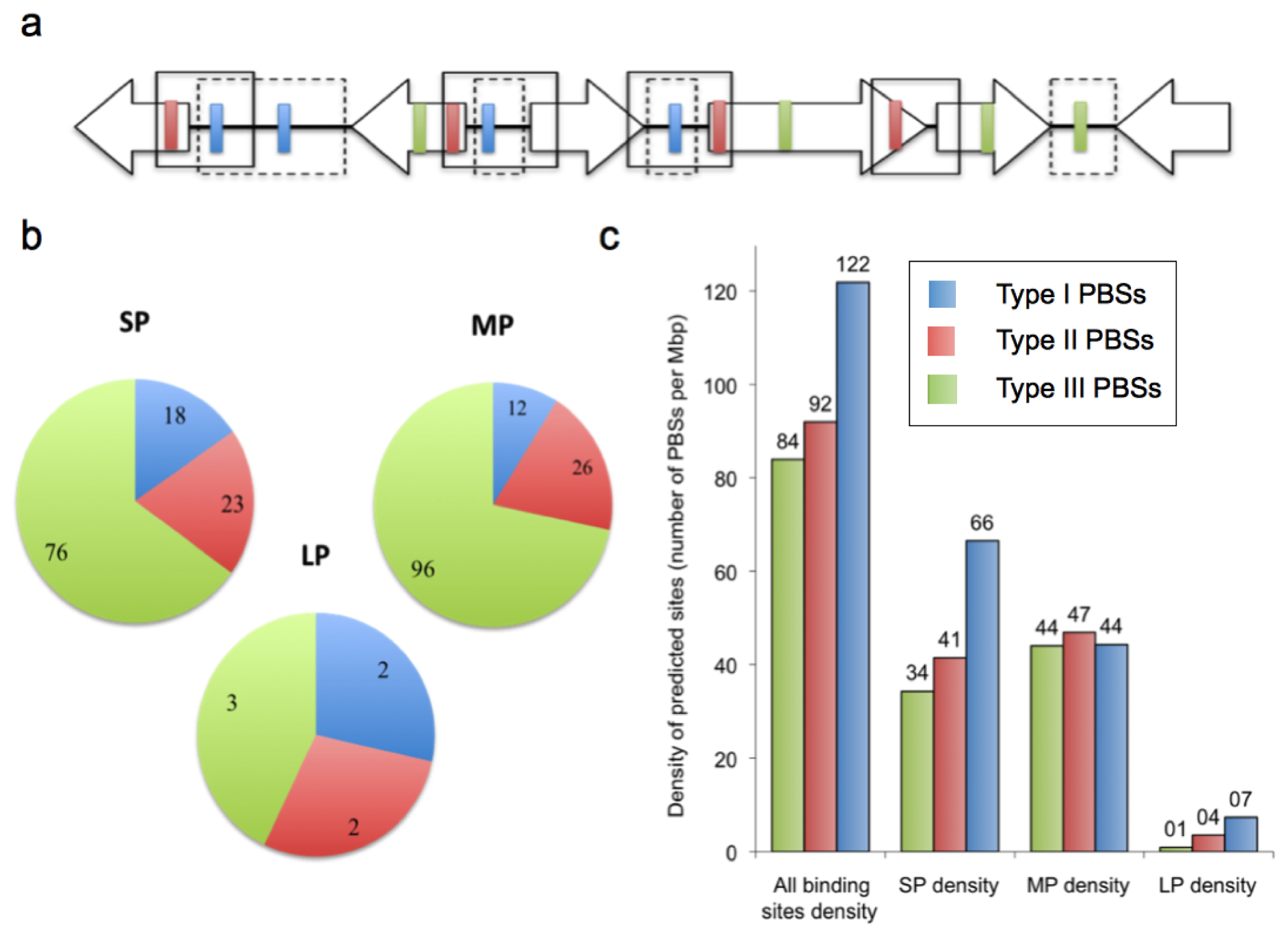
2.2. Prediction of AfeR Binding Sites in the At. ferrooxidans ATCC 23270 Genome
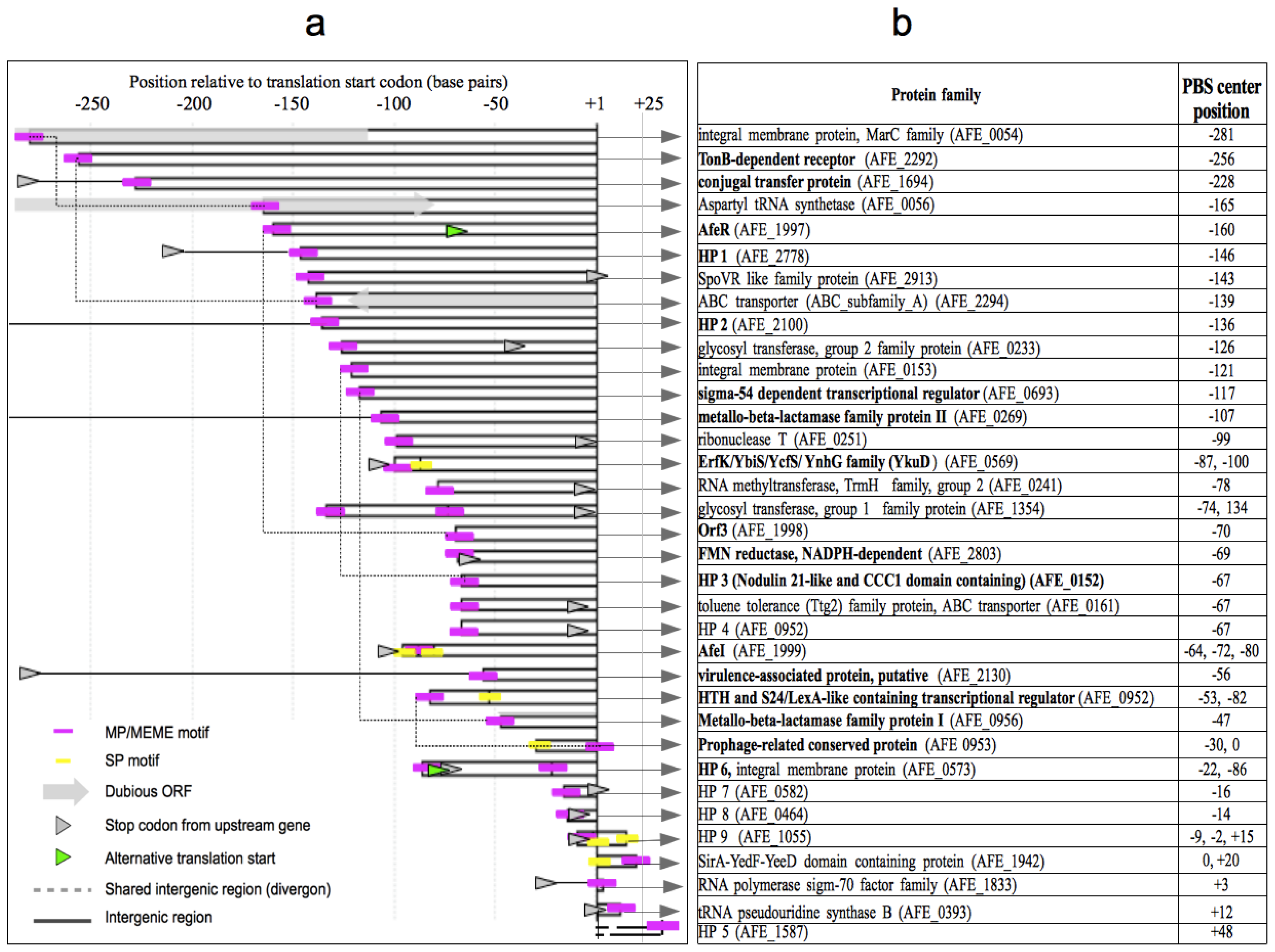
2.3. Identification of Putative Gene Functions Regulated by AfeR
2.4. The At. ferrooxidans Putative Quorum Sensing Regulon
3. Experimental Section
3.1. Construction of Hidden Markov Models (HMMs)
3.2. Analysis of the Genomic Contexts of the Putative AfeR Binding Sites
4. Conclusions
Supplementary Information
ijms-14-16901-s001.pdfAcknowledgments
Conflicts of Interest
References
- Baker, B.J.; Banfield, J.F. Microbial communities in acid mine drainage. FEMS Microbiol. Ecol 2003, 44, 139–152. [Google Scholar]
- Rawlings, D.E.; Johnson, D.B. The microbiology of biomining: Development and optimization of mineral-oxidizing microbial consortia. Microbiology 2007, 153, 315–324. [Google Scholar]
- Rohwerder, T.; Gehrke, T.; Kinzler, K.; Sand, W. Bioleaching review part A. Progress in bioleaching: Fundamentals and mechanisms of bacterial metal sulfide oxidation. Appl. Microbiol. Biotechnol 2003, 63, 239–248. [Google Scholar]
- Sand, W.; Gehrke, T. Extracellular polymeric substances mediate bioleaching/biocorrosion via interfacial processes involving iron(III) ions and acidophilic bacteria. Res. Microbiol 2006, 157, 49–56. [Google Scholar]
- Von Bodman, S.B.; Willey, J.M.; Diggle, S.P. Cell-cell communication in bacteria: United we stand. J. Bacteriol 2008, 190, 4377–4391. [Google Scholar]
- Pappas, K.M.; Weingart, C.L.; Winans, S.C. Chemical communication in proteobacteria: Biochemical and structural studies of signal synthases and receptors required for intercellular signalling. Mol. Microbiol 2004, 53, 755–769. [Google Scholar]
- Valenzuela, S.; Banderas, A.; Jerez, C.A.; Guiliani, N. Cell-cell communication in bacteria: A promising new approach to improve bioleaching efficiency? In Microbial Processing of Metal Sulphides; Sand, W., Donati, E., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 253–254. [Google Scholar]
- Whitehead, N.A.; Barnard, A.M.; Slater, H.; Simpson, N.J.; Salmond, G.P. Quorum-sensing in gram-negative bacteria. FEMS Microbiol. Rev 2001, 25, 365–404. [Google Scholar]
- Daniels, R.; Reynaert, S.; Hoekstra, H.; Verreth, C.; Janssens, J.; Braeken, K.; Fauvart, M.; Beullens, S.; Heusdens, C.; Lambrichts, I.; et al. Quorum signal molecules as biosurfactants affecting swarming in Rhizobium etli. Proc. Natl. Acad. Sci. USA 2006, 103, 14965–14970. [Google Scholar]
- Dubern, J.F.; Lugtenberg, B.J.; Bloemberg, G.V. The ppuI-rsaL-ppuR quorum-sensing system regulates biofilm formation of Pseudomonas putida PCL1445 by controlling biosynthesis of the cyclic lipopeptides putisolvins I and II. J. Bacteriol 2006, 188, 2898–2906. [Google Scholar]
- Labbate, M.; Zhu, H.; Thung, L.; Bandara, R.; Larsen, M.R.; Willcox, M.D.; Givskov, M.; Rice, S.A.; Kjelleberg, S. Quorum-sensing regulation of adhesion in Serratia marcescens MG1 is surface dependent. J. Bacteriol 2007, 189, 2702–2711. [Google Scholar]
- Farah, C.; Vera, M.; Morin, D.; Haras, D.; Jerez, C.A.; Guiliani, N. Evidence for a functional quorum-sensing type AI-1 system in the extremophilic bacterium Acidithiobacillus ferrooxidans. Appl. Environ. Microbiol 2005, 71, 7033–7040. [Google Scholar]
- Rivas, M.; Seeger, M.; Holmes, D.S.; Jedlicki, E. A lux-like quorum sensing system in the extreme acidophile Acidithiobacillus ferrooxidans. Biol. Res 2005, 38, 283–297. [Google Scholar]
- González, A.; Bellenberg, S.; Mamani, S.; Ruiz, L.; Echeverría, A.; Soulère, L.; Doutheau, A.; Demergasso, C.; Sand, W.; Queneau, Y.; et al. AHL signaling molecules with a large acyl chain enhance biofilm formation on sulfur and metal sulfides by the bioleaching bacterium Acidithiobacillus ferrooxidans. Appl. Microbiol. Biotechnol 2013, 97, 3729–3737. [Google Scholar]
- Chambers, C.E.; Lutter, E.I.; Visser, M.B.; Law, P.P.; Sokol, P.A. Identification of potential cepr regulated genes using a cep-box motif-based search of the Burkholderia cenocepacia genome. BMC Microbiol 2006, 6, 104. [Google Scholar]
- Mullapudi, N.; Joseph, S.; Kissinger, J. Identification and functional characterization of cis-regulatory elements in the apicomplexan parasite Toxoplasma gondii. Genome Biol 2009, 10, r34. [Google Scholar]
- Bailey, T.L.; Elkan, C. Fitting a Mixture Model by Expectation Maximization to Discover Motifs in Biopolymers. Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology, Stanford, CA, USA, 14–17 August 1994.Altman, R.; Brutlag, D.; Karp, P.; Lathrop, R.; Searls, D. (Eds.) AAAI Press: Menlo Park, CA, USA, 1994; pp. 28–36.
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 1998, 16, 10881–10890. [Google Scholar]
- Bailey, T.L.; Gribskov, M. Combining evidence using p-values: Application to sequence homology searches. Bioinformatics 1998, 14, 48–54. [Google Scholar]
- Schuster, M.; Urbanowski, M.L.; Greenberg, E.P. Promoter specificity in Pseudomonas aeruginosa quorum sensing revealed by DNA binding of purified LasR. Proc. Natl. Acad. Sci. USA 2004, 101, 15833–15839. [Google Scholar]
- Lavrrar, J.L.; McIntosh, M.A. Architecture of a fur binding site: A comparative analysis. J. Bacteriol 2003, 185, 2194–2202. [Google Scholar]
- Reverchon, S.; Bouillant, M.L.; Salmond, G.; Nasser, W. Integration of the quorum-sensing system in the regulatory networks controlling virulence factor synthesis in Erwinia chrysanthemi. Mol. Microbiol 1998, 29, 1407–1418. [Google Scholar]
- Chatterjee, J.; Miyamoto, C.M.; Zouzoulas, A.; Lang, B.F.; Skouris, N.; Meighen, E.A. MetR and CRP bind to the Vibrio harveyi lux promoters and regulate luminescence. Mol. Microbiol 2002, 46, 101–111. [Google Scholar]
- Rampioni, G.; Bertani, I.; Zennaro, E.; Polticelli, F.; Venturi, V.; Leoni, L. The quorum-sensing negative regulator RsaL of Pseudomonas aeruginosa binds to the lasI promoter. J. Bacteriol 2006, 188, 815–819. [Google Scholar]
- Anderson, R.M.; Zimprich, C.A.; Rust, L. A second operator is involved in Pseudomonas aeruginosa elastase (lasB) activation. J. Bacteriol 1999, 181, 6264–6270. [Google Scholar]
- Pessi, G.; Haas, D. Transcriptional control of the hydrogen cyanide biosynthetic genes hcnABC by the anaerobic regulator ANR and the quorum-sensing regulators lasR and rhlR in Pseudomonas aeruginosa. J. Bacteriol 2000, 182, 6940–6949. [Google Scholar]
- Xiao, G.; He, J.; Rahme, L.G. Mutation analysis of the Pseudomonas aeruginosa mvfR and pqsABCDE gene promoters demonstrates complex quorum-sensing circuitry. Microbiology 2001, 152, 1679–1686. [Google Scholar]
- Andersson, R.A.; Eriksson, A.R.; Heikinheimo, R.; Mae, A.; Pirhonen, M.; Koiv, V.; Hyytiainen, H.; Tuikkala, A.; Palva, E.T. Quorum sensing in the plant pathogen Erwinia carotovora subsp. carotovora: The role of expR(cc). Mol. Plant Microbe Interact 2000, 13, 384–393. [Google Scholar]
- Minogue, T.D.; Wehland-von Trebra, M.; Bernhard, F.; von Bodman, S.B. The autoregulatory role of EsaR, a quorum-sensing regulator in Pantoea stewartii ssp. stewartii: Evidence for a repressor function. Mol. Microbiol 2002, 44, 1625–1635. [Google Scholar]
- Von Bodman, S.B.; Ball, J.K.; Faini, M.A.; Herrera, C.M.; Minogue, T.D.; Urbanowski, M.L.; Stevens, A.M. The quorum sensing negative regulators EsaR and ExpR(ecc), homologues within the LuxR family, retain the ability to function as activators of transcription. J. Bacteriol 2003, 185, 7001–7007. [Google Scholar]
- Whiteley, M.; Greenberg, E.P. Promoter specificity elements in pseudomonas aeruginosa quorum-sensing-controlled genes. J. Bacteriol 2001, 183, 5529–5534. [Google Scholar]
- Khan, S.R.; Mavrodi, D.V.; Jog, G.J.; Suga, H.; Thomashow, L.S.; Farrand, S.K. Activation of the phz operon of Pseudomonas fluorescens 2-79 requires the LuxR homolog PhzR, N-(3-OH-hexanoyl)-L-homoserine lactone produced by the LuxI homolog PhzI, and a cis-acting phz box. J. Bacteriol 2005, 187, 6517–6527. [Google Scholar]
- Dong, Y.H.; Zhang, L.H. Quorum sensing and quorum-quenching enzymes. J. Microbiol 2005, 43, 101–109. [Google Scholar]
- Abbas, A.; Adams, C.; Scully, N.; Glennon, J.; Ogara, F. A role for TonB1 in biofilm formation and quorum sensing in Pseudomonas aeruginosa. FEMS Microbiol. Lett 2007, 274, 269–278. [Google Scholar]
- Motorin, Y.; Helm, M. RNA nucleotide methylation. Wiley Interdiscip. Rev. RNA 2011, 2, 611–631. [Google Scholar]
- Hwang, I.; Smyth, A.J.; Luo, Z.Q.; Farrand, S.K. Modulating quorum sensing by antiactivation: TraM interacts with TraR to inhibit activation of Ti plasmid conjugal transfer genes. Mol. Microbiol 1999, 34, 282–294. [Google Scholar]
- Ramos, A.; Boels, I.C.; de Vos, W.M.; Santos, H. Relationship between glycolysis and exopolysaccharide biosynthesis in Lactococcus lactis. Appl. Environ. Microbiol 2001, 67, 33–41. [Google Scholar]
- Silo-Suh, L.; Suh, S.J.; Phibbs, P.V.; Ohman, D.E. Adaptations of Pseudomonas aeruginosa to the cystic fibrosis lung environment can include deregulation of zwf, encoding glucose-6-phosphate dehydrogenase. J. Bacteriol 2005, 187, 7561–7568. [Google Scholar]
- Barreto, M.; Jedlicki, E.; Holmes, D.S. Identification of a gene cluster for the formation of extracellular polysaccharide precursors in the chemolithoautotroph Acidithiobacillus ferrooxidans. Appl. Environ. Microbiol 2005, 71, 2902–2909. [Google Scholar]
- Arredondo, R.; García, A.; Jerez, C.A. Partial removal of lipopolysaccharide from Thiobacillus ferrooxidans affects its adhesion to solids. Appl. Environ. Microbiol 1994, 60, 2846–2851. [Google Scholar]
- Gehrke, T.; Hallmann, R.; Kinzler, K.; Sand, W. The EPS of Acidithiobacillus ferrooxidans: A model for structure-function relationships of attached bacteria and their physiology. Water Sci. Technol 2001, 43, 159–167. [Google Scholar]
- Mamani, S.; Denis, Y.; Moinier, D.; Sabbah, M.; Soulère, L.; Queneau, Y.; Bonnefoy, V.; Guiliani, N. Characterization of the quorum sensing regulon in Acidithiobacillus ferrooxidans. Adv. Mat. Res. 2013, in press. [Google Scholar]
- Sabbah, M.; Fontaine, F.; Grand, L.; Boukraa, M.; Efrit, M.L.; Doutheau, A.; Soulère, L.; Queneau, Y. Synthesis and biological evaluation of new N-acyl-homoserine-lactone analogues, based on triazole and tetrazole scaffolds, acting as LuxR-dependent quorum sensing modulators. Bioorg. Med. Chem 2012, 20, 4727–4736. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. Clustal W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994, 22, 4673–4680. [Google Scholar]
- Eddy, S.R. Profile hidden markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res 1997, 25, 3389–3402. [Google Scholar]
- Marchler-Bauer, A.; Bryant, S.H. CD-search: Protein domain annotations on the fly. Nucleic Acids Res 2004, 32, w327–w331. [Google Scholar]
- Wernersson, R. FeatureExtract-Extraction of sequence annotation made easy. Nucleic Acids Res 2005, 33, w567–w569. [Google Scholar]
- Stothard, P. The sequence manipulation suite: Javascript programs for analyzing and formatting protein and DNA sequences. Biotechniques 2000, 28, 1102–1104. [Google Scholar]
- Villesen, P. FaBox: An online toolbox for fasta sequences. Mol. Ecol. Notes 2007, 7, 965–968. [Google Scholar]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res 2006, 34, w369–w373. [Google Scholar]
- Schuster, M.; Lostroh, C.P.; Ogi, T.; Greenberg, E.P. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: A transcriptome analysis. J. Bacteriol 2003, 185, 2066–2079. [Google Scholar]
- Vera, M.; Krok, B.; Bellenberg, S.; Sand, W.; Poetsch, A. Shotgun proteomics study of early biofilm formation process of Acidithiobacillus ferrooxidans ATCC 23270 on pyrite. Proteomics 2013, 13, 1133–1144. [Google Scholar]




© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Banderas, A.; Guiliani, N. Bioinformatic Prediction of Gene Functions Regulated by Quorum Sensing in the Bioleaching Bacterium Acidithiobacillus ferrooxidans. Int. J. Mol. Sci. 2013, 14, 16901-16916. https://doi.org/10.3390/ijms140816901
Banderas A, Guiliani N. Bioinformatic Prediction of Gene Functions Regulated by Quorum Sensing in the Bioleaching Bacterium Acidithiobacillus ferrooxidans. International Journal of Molecular Sciences. 2013; 14(8):16901-16916. https://doi.org/10.3390/ijms140816901
Chicago/Turabian StyleBanderas, Alvaro, and Nicolas Guiliani. 2013. "Bioinformatic Prediction of Gene Functions Regulated by Quorum Sensing in the Bioleaching Bacterium Acidithiobacillus ferrooxidans" International Journal of Molecular Sciences 14, no. 8: 16901-16916. https://doi.org/10.3390/ijms140816901
APA StyleBanderas, A., & Guiliani, N. (2013). Bioinformatic Prediction of Gene Functions Regulated by Quorum Sensing in the Bioleaching Bacterium Acidithiobacillus ferrooxidans. International Journal of Molecular Sciences, 14(8), 16901-16916. https://doi.org/10.3390/ijms140816901