Egr-1 Upregulates Siva-1 Expression and Induces Cardiac Fibroblast Apoptosis

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
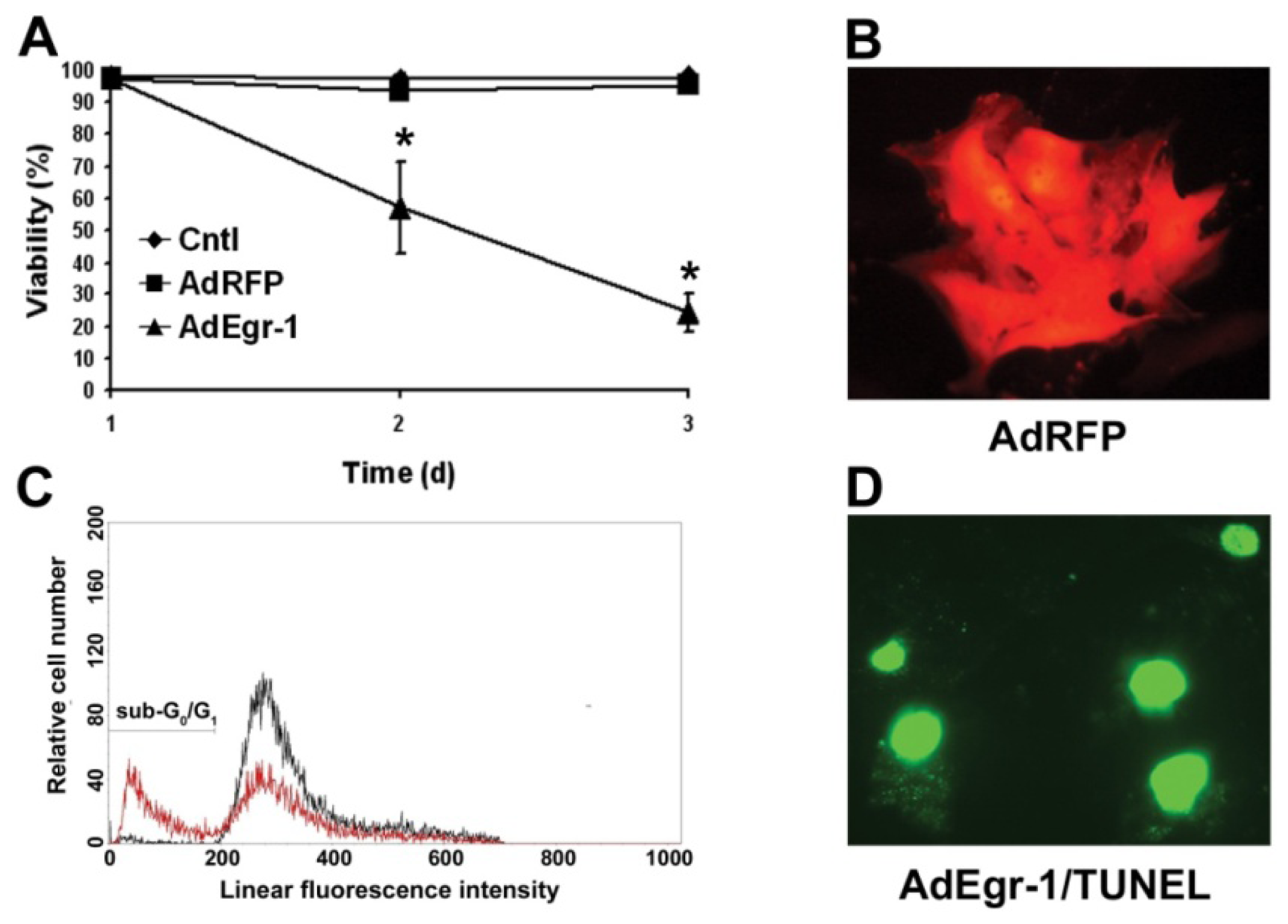
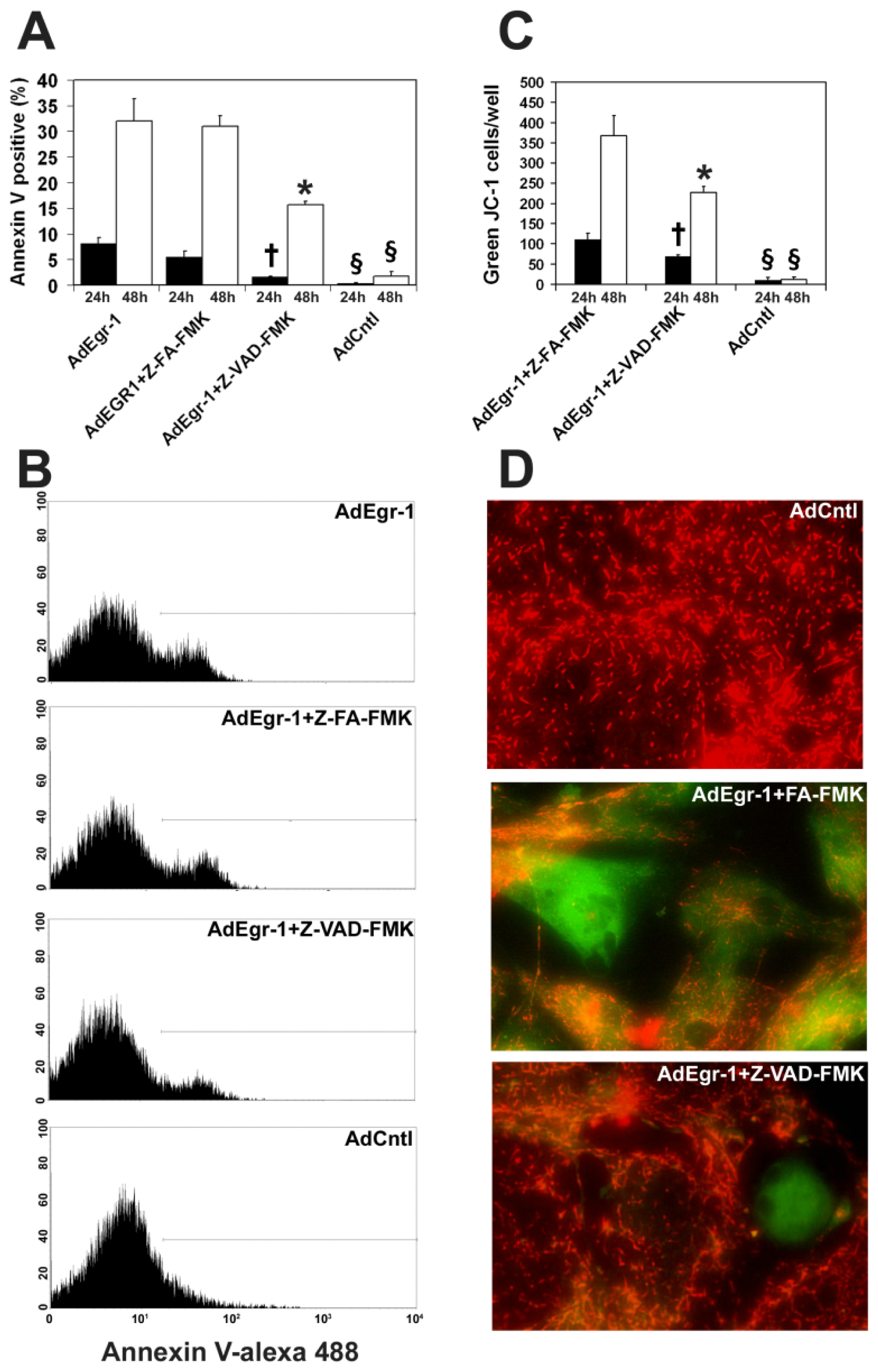
2.1.1. Egr-1 Induces Caspase Sensitive Cardiac Fibroblast Apoptosis in Vitro
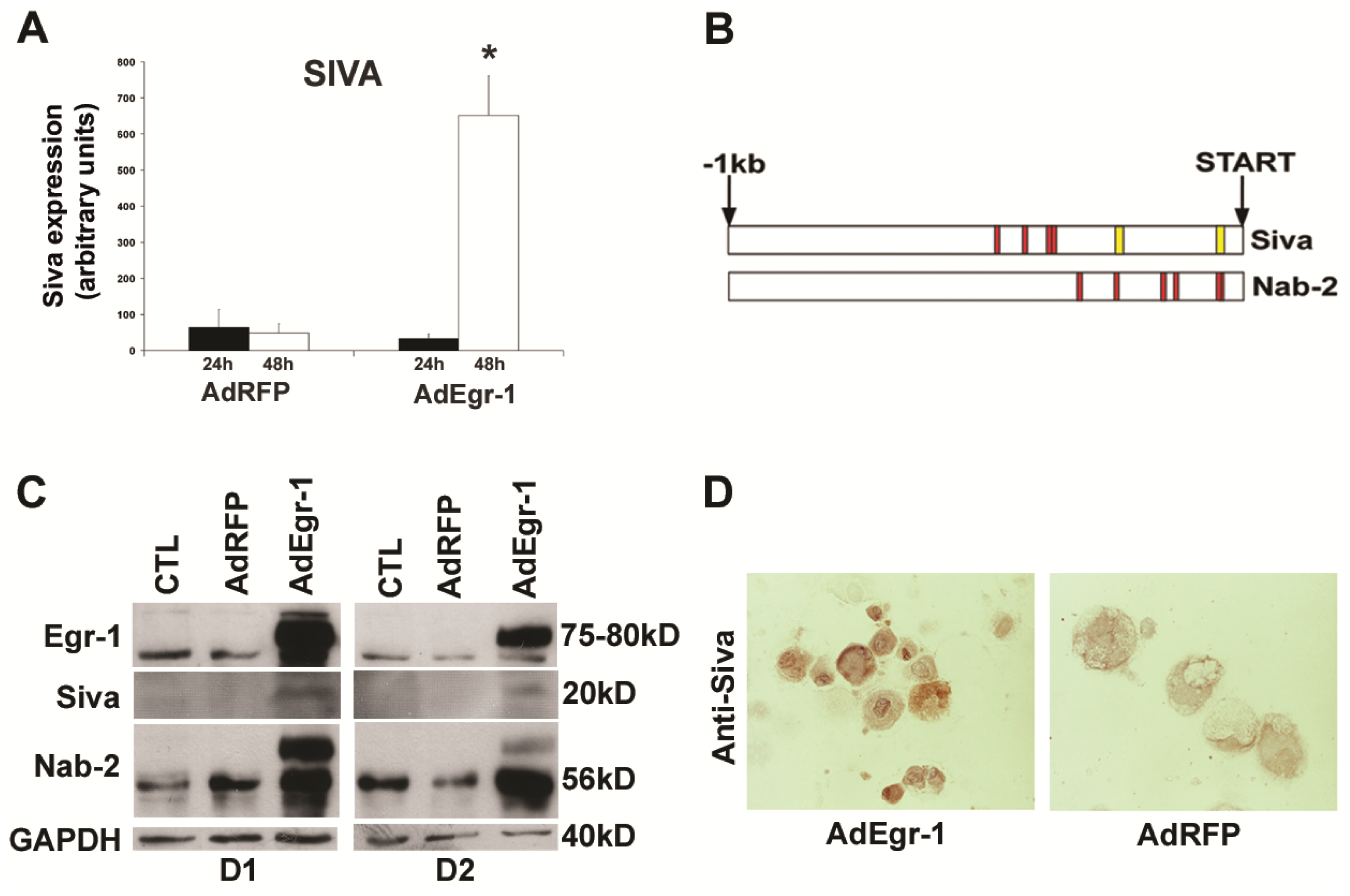
2.1.2. Egr-1 Mediated Transcriptional Regulation during Cardiac Fibroblast Apoptosis
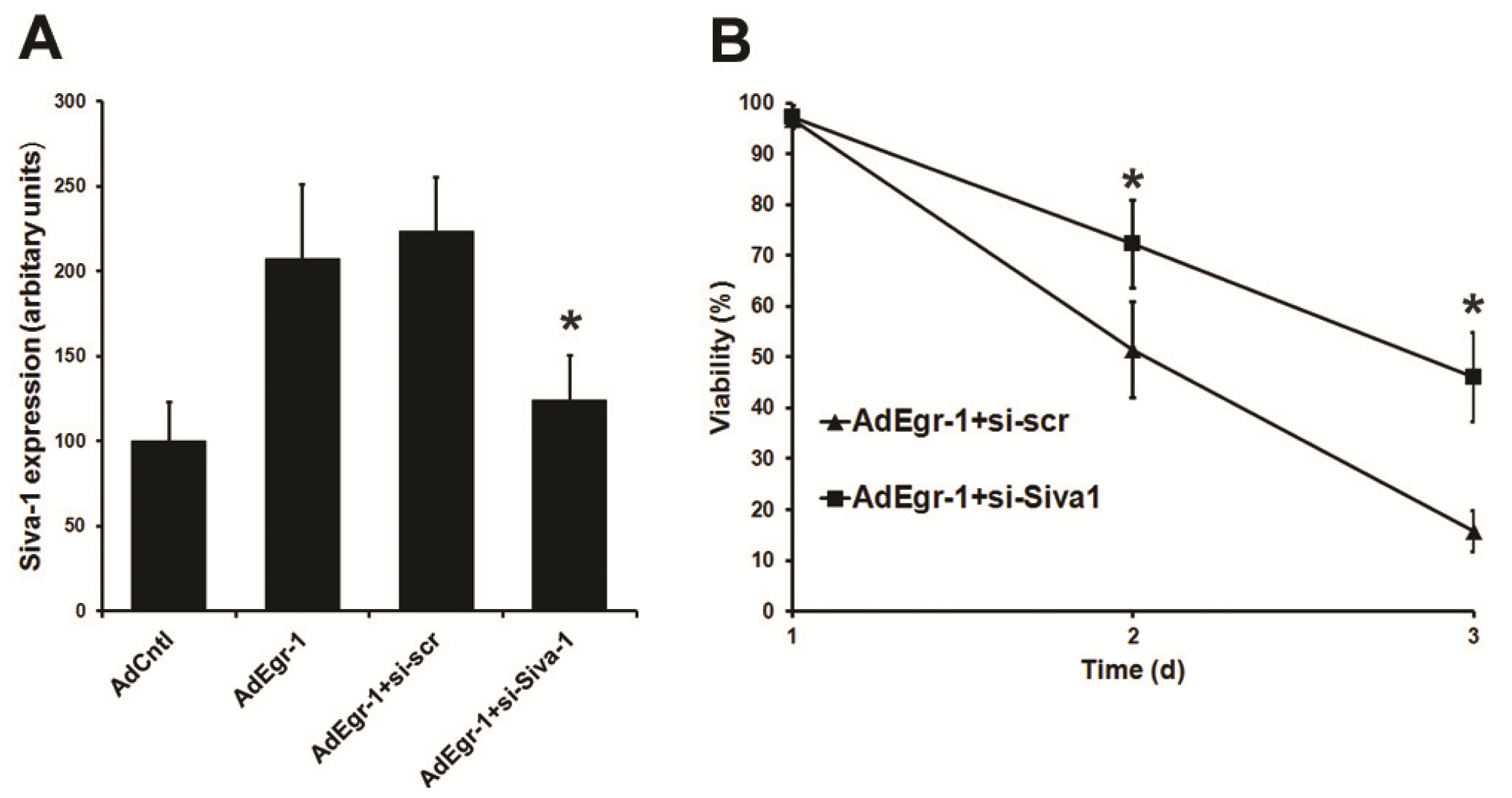
2.1.3. Suppression of Siva-1 Expression Reduces Egr-1 Mediated Cardiac Fibroblast Cell Death
2.2. Discussion
3. Experimental Section
3.2. Culture, Infection and Transfection of Cardiac Fibroblasts
3.3. Assays of Viability and Cell Death
3.4. Promoter Analysis
3.5. Real Time RT-PCR Analysis
3.6. Western Blotting and Histochemistry
3.7. Data Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther 2009, 123, 255–278. [Google Scholar]
- Brown, R.D.; Ambler, S.K.; Mitchell, M.D.; Long, C.S. The cardiac fibroblast: Therapeutic target in myocardial remodeling and failure. Annu. Rev. Pharmacol. Toxicol 2005, 45, 657–687. [Google Scholar]
- Weber, K.T. Fibrosis in hypertensive heart disease: Focus on cardiac fibroblasts. J. Hypertens 2004, 22, 47–50. [Google Scholar]
- Baudino, T.A.; Carver, W.; Giles, W.; Borg, T.K. Cardiac fibroblasts: Friend or foe? Am. J. Physiol. Heart Circ. Physiol 2006, 291, H1015–H1026. [Google Scholar]
- Souders, C.A.; Bowers, S.L.; Baudino, T.A. Cardiac fibroblast: The renaissance cell. Circ. Res 2009, 105, 1164–1176. [Google Scholar]
- Takano, H.; Hasegawa, H.; Nagai, T.; Komuro, I. Implication of cardiac remodeling in heart failure: Mechanisms and therapeutic strategies. Intern. Med 2003, 42, 465–469. [Google Scholar]
- Spinale, F.G. Myocardial matrix remodeling and the matrix metalloproteinases: Influence on cardiac form and function. Physiol. Rev 2007, 87, 1285–1342. [Google Scholar]
- Der Sarkissian, S.; Marchand, E.L.; Duguay, D.; Hamet, P.; de Blois, D. Reversal of interstitial fibroblast hyperplasia via apoptosis in hypertensive rat heart with valsartan or enalapril. Cardiovasc. Res 2003, 57, 775–783. [Google Scholar]
- Duguay, D.; Pesant, S.; Deschepper, C.F.; deBlois, D. Fibroblast apoptosis precedes cardiomyocyte mass reduction during left ventricular remodeling in hypertensive rats treated with amlodipine. J. Hypertens 2007, 25, 1291–1299. [Google Scholar]
- Takeda, N.; Manabe, I.; Uchino, Y.; Eguchi, K.; Matsumoto, S.; Nishimura, S.; Shindo, T.; Sano, M.; Otsu, K.; Snider, P.; et al. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J. Clin. Invest 2010, 120, 254–265. [Google Scholar]
- Creemers, E.E.; Pinto, Y.M. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc. Res 2011, 89, 265–272. [Google Scholar]
- Tijsen, A.J.; Pinto, Y.M.; Creemers, E.E. Non-cardiomyocyte microRNAs in heart failure. Cardiovasc. Res 2012, 93, 573–582. [Google Scholar]
- Kumar, S. Caspase function in programmed cell death. Cell Death Differ 2007, 14, 32–43. [Google Scholar]
- Logue, S.E.; Martin, S.J. Caspase activation cascades in apoptosis. Biochem. Soc. Trans 2008, 36, 1–9. [Google Scholar]
- Kumar, S.; Cakouros, D. Transcriptional control of the core cell-death machinery. Trends Biochem. Sci 2004, 29, 193–199. [Google Scholar]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar]
- Gustafsson, A.B.; Gottlieb, R.A. Mechanisms of apoptosis in the heart. J. Clin. Immunol 2003, 23, 447–459. [Google Scholar]
- Saraste, A.; Pulkki, K.; Kallajoki, M.; Heikkila, P.; Laine, P.; Mattila, S.; Nieminen, M.S.; Parvinen, M.; Voipio-Pulkki, L.M. Cardiomyocyte apoptosis and progression of heart failure to transplantation. Eur. J. Clin. Invest 1999, 29, 380–386. [Google Scholar]
- Takemura, G.; Ohno, M.; Hayakawa, Y.; Misao, J.; Kanoh, M.; Ohno, A.; Uno, Y.; Minatoguchi, S.; Fujiwara, T.; Fujiwara, H. Role of apoptosis in the disappearance of infiltrated and proliferated interstitial cells after myocardial infarction. Circ. Res 1998, 82, 1130–1138. [Google Scholar]
- Park, M.; Shen, Y.T.; Gaussin, V.; Heyndrickx, G.R.; Bartunek, J.; Resuello, R.R.; Natividad, F.F.; Kitsis, R.N.; Vatner, D.E.; Vatner, S.F. Apoptosis predominates in nonmyocytes in heart failure. Am. J. Physiol. Heart Circ. Physiol 2009, 297, H785–H791. [Google Scholar]
- Milbrandt, J. A nerve growth factor-induced gene encodes a possible transcriptional regulatory factor. Science 1987, 238, 797–799. [Google Scholar]
- Bhattacharyya, S.; Fang, F.; Tourtellotte, W.; Varga, J. Egr-1: New conductor for the tissue repair orchestra directs harmony (regeneration) or cacophony (fibrosis). J. Pathol 2013, 229, 286–297. [Google Scholar]
- Thiel, G.; Cibelli, G. Regulation of life and death by the zinc finger transcription factor Egr-1. J. Cell. Physiol 2002, 193, 287–292. [Google Scholar]
- Fu, J.D.; Stone, N.R.; Liu, L.; Spencer, C.I.; Qian, L.; Hayashi, Y.; Delgado-Olguin, P.; Ding, S.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of human fibroblasts toward a cardiomyocyte-like state. Stem Cell Reports 2013, 1, 235–247. [Google Scholar]
- Christy, B.A.; Lau, L.F.; Nathans, D. A gene activated in mouse 3T3 cells by serum growth factors encodes a protein with “zinc finger” sequences. Proc. Natl. Acad. Sci. USA 1988, 85, 7857–7861. [Google Scholar]
- Lim, R.W.; Varnum, B.C.; O’Brien, T.G.; Herschman, H.R. Induction of tumor promotor-inducible genes in murine 3T3 cell lines and tetradecanoyl phorbol acetate-nonproliferative 3T3 variants can occur through protein kinase C-dependent and -independent pathways. Mol. Cell. Biol 1989, 9, 1790–1793. [Google Scholar]
- Lemaire, P.; Vesque, C.; Schmitt, J.; Stunnenberg, H.; Frank, R.; Charnay, P. The serum-inducible mouse gene Krox-24 encodes a sequence-specific transcriptional activator. Mol. Cell. Biol 1990, 10, 3456–3467. [Google Scholar]
- Lucerna, M.; Pomyje, J.; Mechtcheriakova, D.; Kadl, A.; Gruber, F.; Bilban, M.; Sobanov, Y.; Schabbauer, G.; Breuss, J.; Wagner, O.; et al. Sustained expression of early growth response protein-1 blocks angiogenesis and tumor growth. Cancer Res 2006, 66, 6708–6713. [Google Scholar]
- Fu, M.; Zhu, X.; Zhang, J.; Liang, J.; Lin, Y.; Zhao, L.; Ehrengruber, M.U.; Chen, Y.E. Egr-1 target genes in human endothelial cells identified by microarray analysis. Gene 2003, 315, 33–41. [Google Scholar]
- Swirnoff, A.H.; Milbrandt, J. DNA-binding specificity of NGFI-A and related zinc finger transcription factors. Mol. Cell. Biol 1995, 15, 2275–2287. [Google Scholar]
- Christy, B.; Nathans, D. DNA binding site of the growth factor-inducible protein Zif268. Proc. Natl. Acad. Sci. USA 1989, 86, 8737–8741. [Google Scholar]
- Cao, X.; Mahendran, R.; Guy, G.R.; Tan, Y.H. Detection and characterization of cellular EGR-1 binding to its recognition site. J. Biol. Chem 1993, 268, 16949–16957. [Google Scholar]
- Svaren, J.; Sevetson, B.R.; Apel, E.D.; Zimonjic, D.B.; Popescu, N.C.; Milbrandt, J. NAB2, a corepressor of NGFI-A (Egr-1) and Krox20, is induced by proliferative and differentiative stimuli. Mol. Cell. Biol 1996, 16, 3545–3553. [Google Scholar]
- Kumbrink, J.; Gerlinger, M.; Johnson, J.P. Egr-1 induces the expression of its corepressor Nab2 by activation of the Nab2 promoter thereby establishing a negative feedback loop. J. Biol. Chem 2005, 280, 42785–42793. [Google Scholar]
- Khachigian, L.M. Early growth response-1 in cardiovascular pathobiology. Circ. Res 2006, 98, 186–191. [Google Scholar]
- Rayner, B.S.; Figtree, G.A.; Sabaretnam, T.; Shang, P.; Mazhar, J.; Weaver, J.C.; Lay, W.N.; Witting, P.K.; Hunyor, S.N.; Grieve, S.M.; et al. Selective inhibition of the master regulator transcription factor Egr-1 with catalytic oligonucleotides reduces myocardial injury and improves left ventricular systolic function in a preclinical model of myocardial infarction. J. Am. Heart Assoc 2013, 2, e000023. [Google Scholar]
- Harja, E.; Bucciarelli, L.G.; Lu, Y.; Stern, D.M.; Zou, Y.S.; Schmidt, A.M.; Yan, S.F. Early growth response-1 promotes atherogenesis: Mice deficient in early growth response-1 and apolipoprotein E display decreased atherosclerosis and vascular inflammation. Circ. Res 2004, 94, 333–339. [Google Scholar]
- Fahmy, R.G.; Dass, C.R.; Sun, L.Q.; Chesterman, C.N.; Khachigian, L.M. Transcription factor Egr-1 supports FGF-dependent angiogenesis during neovascularization and tumor growth. Nat. Med 2003, 9, 1026–1032. [Google Scholar]
- Arai, M.; Yoguchi, A.; Takizawa, T.; Yokoyama, T.; Kanda, T.; Kurabayashi, M.; Nagai, R. Mechanism of doxorubicin-induced inhibition of sarcoplasmic reticulum Ca2+-ATPase gene transcription. Circ. Res 2000, 86, 8–14. [Google Scholar]
- Bhindi, R.; Khachigian, L.M.; Lowe, H.C. DNAzymes targeting the transcription factor Egr-1 reduce myocardial infarct size following ischemia-reperfusion in rats. J. Thromb. Haemost 2006, 4, 1479–1483. [Google Scholar]
- Kwon, O.; Soung, N.K.; Thimmegowda, N.R.; Jeong, S.J.; Jang, J.H.; Moon, D.O.; Chung, J.K.; Lee, K.S.; Kwon, Y.T.; Erikson, R.L.; et al. Patulin induces colorectal cancer cells apoptosis through EGR-1 dependent ATF3 up-regulation. Cell Signal 2012, 24, 943–950. [Google Scholar]
- Deblois, D.; Tea, B.S.; Beaudry, D.; Hamet, P. Regulation of therapeutic apoptosis: A potential target in controlling hypertensive organ damage. Can. J. Physiol. Pharmacol 2005, 83, 29–41. [Google Scholar]
- Henke, A.; Launhardt, H.; Klement, K.; Stelzner, A.; Zell, R.; Munder, T. Apoptosis in coxsackievirus B3-caused diseases: Interaction between the capsid protein VP2 and the proapoptotic protein siva. J. Virol 2000, 74, 4284–4290. [Google Scholar]
- Smiley, S.T.; Reers, M.; Mottola-Hartshorn, C.; Lin, M.; Chen, A.; Smith, T.W.; Steele, G.D., Jr.; Chen, L.B. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc. Natl. Acad. Sci. USA 1991, 88, 3671–3675. [Google Scholar]
- Prasad, K.V.; Ao, Z.; Yoon, Y.; Wu, M.X.; Rizk, M.; Jacquot, S.; Schlossman, S.F. CD27, a member of the tumor necrosis factor receptor family, induces apoptosis and binds to Siva, a proapoptotic protein. Proc. Natl. Acad. Sci. USA 1997, 94, 6346–6351. [Google Scholar]
- Py, B.; Slomianny, C.; Auberger, P.; Petit, P.X.; Benichou, S. Siva-1 and an alternative splice form lacking the death domain, Siva-2, similarly induce apoptosis in T lymphocytes via a caspase-dependent mitochondrial pathway. J. Immunol 2004, 172, 4008–4017. [Google Scholar]
- Mei, Y.; Wu, M. Multifaceted functions of Siva-1: More than an Indian god of destruction. Protein Cell 2012, 3, 117–122. [Google Scholar]
- Fortin, A.; MacLaurin, J.G.; Arbour, N.; Cregan, S.P.; Kushwaha, N.; Callaghan, S.M.; Park, D.S.; Albert, P.R.; Slack, R.S. The proapoptotic gene SIVA is a direct transcriptional target for the tumor suppressors p53 and E2F1. J. Biol. Chem 2004, 279, 28706–28714. [Google Scholar]
- Xiao, H.; Palhan, V.; Yang, Y.; Roeder, R.G. TIP30 has an intrinsic kinase activity required for up-regulation of a subset of apoptotic genes. EMBO J 2000, 19, 956–963. [Google Scholar]
- Svaren, J.; Sevetson, B.R.; Golda, T.; Stanton, J.J.; Swirnoff, A.H.; Milbrandt, J. Novel mutants of NAB corepressors enhance activation by Egr transactivators. EMBO J 1998, 17, 6010–6019. [Google Scholar]
- Saadane, N.; Alpert, L.; Chalifour, L.E. Altered molecular response to adrenoreceptor-induced cardiac hypertrophy in Egr-1-deficient mice. Am. J. Physiol. Heart Circ. Physiol 2000, 278, H796–805. [Google Scholar]
- Autieri, M.V.; Kelemen, S.E.; Gaughan, J.P.; Eisen, H.J. Early growth responsive gene (Egr)-1 expression correlates with cardiac allograft rejection. Transplantation 2004, 78, 107–111. [Google Scholar]
- Okada, M.; Wang, C.Y.; Hwang, D.W.; Sakaguchi, T.; Olson, K.E.; Yoshikawa, Y.; Minamoto, K.; Mazer, S.P.; Yan, S.F.; Pinsky, D.J. Transcriptional control of cardiac allograft vasculopathy by early growth response gene-1 (Egr-1). Circ. Res 2002, 91, 135–142. [Google Scholar]
- Zhang, Y.; Shi, G.; Zheng, J.; Tang, Z.; Gao, P.; Lv, Y.; Guo, F.; Jia, Q. The protective effects of N-n-butyl haloperidol iodide on myocardial ischemia-reperfusion injury in rats by inhibiting Egr-1 overexpression. Cell. Physiol. Biochem 2007, 20, 639–648. [Google Scholar]
- Khachigian, L.M. Suppressing a sick heart. Nat. Med 2005, 11, 828–829. [Google Scholar]
- Buitrago, M.; Lorenz, K.; Maass, A.H.; Oberdorf-Maass, S.; Keller, U.; Schmitteckert, E.M.; Ivashchenko, Y.; Lohse, M.J.; Engelhardt, S. The transcriptional repressor Nab1 is a specific regulator of pathological cardiac hypertrophy. Nat. Med 2005, 11, 837–844. [Google Scholar]
- Tsai, J.C.; Liu, L.; Cooley, B.C.; DiChiara, M.R.; Topper, J.N.; Aird, W.C. The Egr-1 promoter contains information for constitutive and inducible expression in transgenic mice. FASEB J 2000, 14, 1870–1872. [Google Scholar]
- Wang, C.; Dostanic, S.; Servant, N.; Chalifour, L.E. Egr-1 negatively regulates expression of the sodium-calcium exchanger-1 in cardiomyocytes in vitro and in vivo. Cardiovasc Res 2005, 65, 187–194. [Google Scholar]
- Lee, S.L.; Tourtellotte, L.C.; Wesselschmidt, R.L.; Milbrandt, J. Growth and differentiation proceeds normally in cells deficient in the immediate early gene NGFI-A. J. Biol. Chem 1995, 270, 9971–9977. [Google Scholar]
- Chu, F.; Borthakur, A.; Sun, X.; Barkinge, J.; Gudi, R.; Hawkins, S.; Prasad, K.V. The Siva-1 putative amphipathic helical region (SAH) is sufficient to bind to BCL-XL and sensitize cells to UV radiation induced apoptosis. Apoptosis 2004, 9, 83–95. [Google Scholar]
- Cao, C.; Ren, X.; Kharbanda, S.; Koleske, A.; Prasad, K.V.; Kufe, D. The ARG tyrosine kinase interacts with Siva-1 in the apoptotic response to oxidative stress. J. Biol. Chem 2001, 276, 11465–11468. [Google Scholar]
- Lucerna, M.; Mechtcheriakova, D.; Kadl, A.; Schabbauer, G.; Schafer, R.; Gruber, F.; Koshelnick, Y.; Muller, H.D.; Issbrucker, K.; Clauss, M.; et al. NAB2, a corepressor of EGR-1, inhibits vascular endothelial growth factor-mediated gene induction and angiogenic responses of endothelial cells. J. Biol. Chem 2003, 278, 11433–11440. [Google Scholar]
- Gomez-Foix, A.M.; Coats, W.S.; Baque, S.; Alam, T.; Gerard, R.D.; Newgard, C.B. Adenovirus-mediated transfer of the muscle glycogen phosphorylase gene into hepatocytes confers altered regulation of glycogen metabolism. J. Biol. Chem 1992, 267, 25129–25134. [Google Scholar]
- Quandt, K.; Frech, K.; Karas, H.; Wingender, E.; Werner, T. MatInd and MatInspector: New fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res 1995, 23, 4878–4884. [Google Scholar]
- Schafer, R.; Abraham, D.; Paulus, P.; Blumer, R.; Grimm, M.; Wojta, J.; Aharinejad, S. Impaired VE-cadherin/β-catenin expression mediates endothelial cell degeneration in dilated cardiomyopathy. Circulation 2003, 108, 1585–1591. [Google Scholar]
- Paulus, P.; Stanley, E.R.; Schafer, R.; Abraham, D.; Aharinejad, S. Colony-stimulating factor-1 antibody reverses chemoresistance in human MCF-7 breast cancer xenografts. Cancer Res 2006, 66, 4349–4356. [Google Scholar]
- Abraham, D.; Hofbauer, R.; Schafer, R.; Blumer, R.; Paulus, P.; Miksovsky, A.; Traxler, H.; Kocher, A.; Aharinejad, S. Selective downregulation of VEGF-A(165), VEGF-R(1), and decreased capillary density in patients with dilative but not ischemic cardiomyopathy. Circ. Res 2000, 87, 644–647. [Google Scholar]




© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zins, K.; Pomyje, J.; Hofer, E.; Abraham, D.; Lucas, T.; Aharinejad, S. Egr-1 Upregulates Siva-1 Expression and Induces Cardiac Fibroblast Apoptosis. Int. J. Mol. Sci. 2014, 15, 1538-1553. https://doi.org/10.3390/ijms15011538
Zins K, Pomyje J, Hofer E, Abraham D, Lucas T, Aharinejad S. Egr-1 Upregulates Siva-1 Expression and Induces Cardiac Fibroblast Apoptosis. International Journal of Molecular Sciences. 2014; 15(1):1538-1553. https://doi.org/10.3390/ijms15011538
Chicago/Turabian StyleZins, Karin, Jiri Pomyje, Erhard Hofer, Dietmar Abraham, Trevor Lucas, and Seyedhossein Aharinejad. 2014. "Egr-1 Upregulates Siva-1 Expression and Induces Cardiac Fibroblast Apoptosis" International Journal of Molecular Sciences 15, no. 1: 1538-1553. https://doi.org/10.3390/ijms15011538
APA StyleZins, K., Pomyje, J., Hofer, E., Abraham, D., Lucas, T., & Aharinejad, S. (2014). Egr-1 Upregulates Siva-1 Expression and Induces Cardiac Fibroblast Apoptosis. International Journal of Molecular Sciences, 15(1), 1538-1553. https://doi.org/10.3390/ijms15011538