Endogenous Nitric-Oxide Synthase Inhibitor ADMA after Acute Brain Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
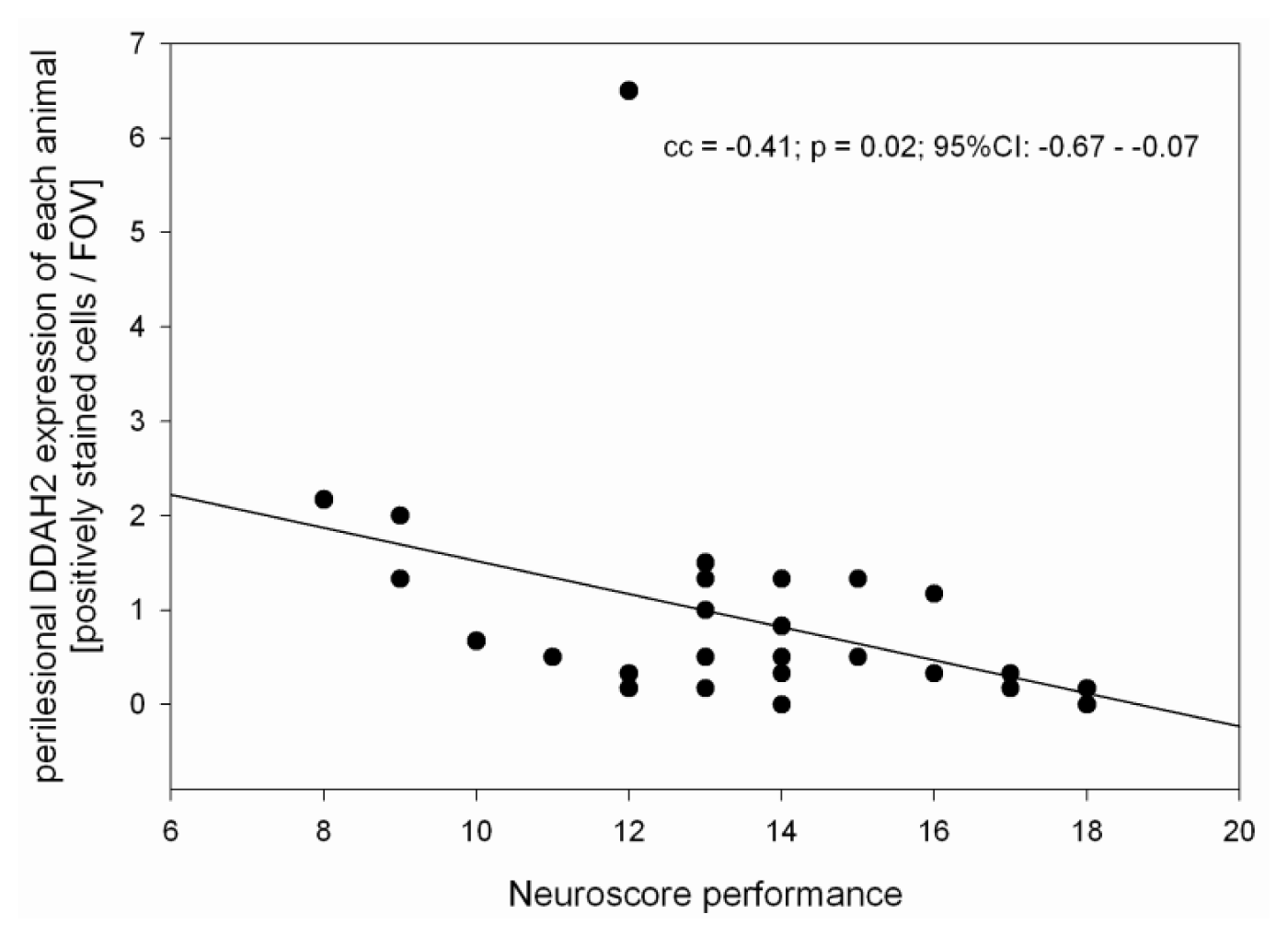
2.1. Neuroscore
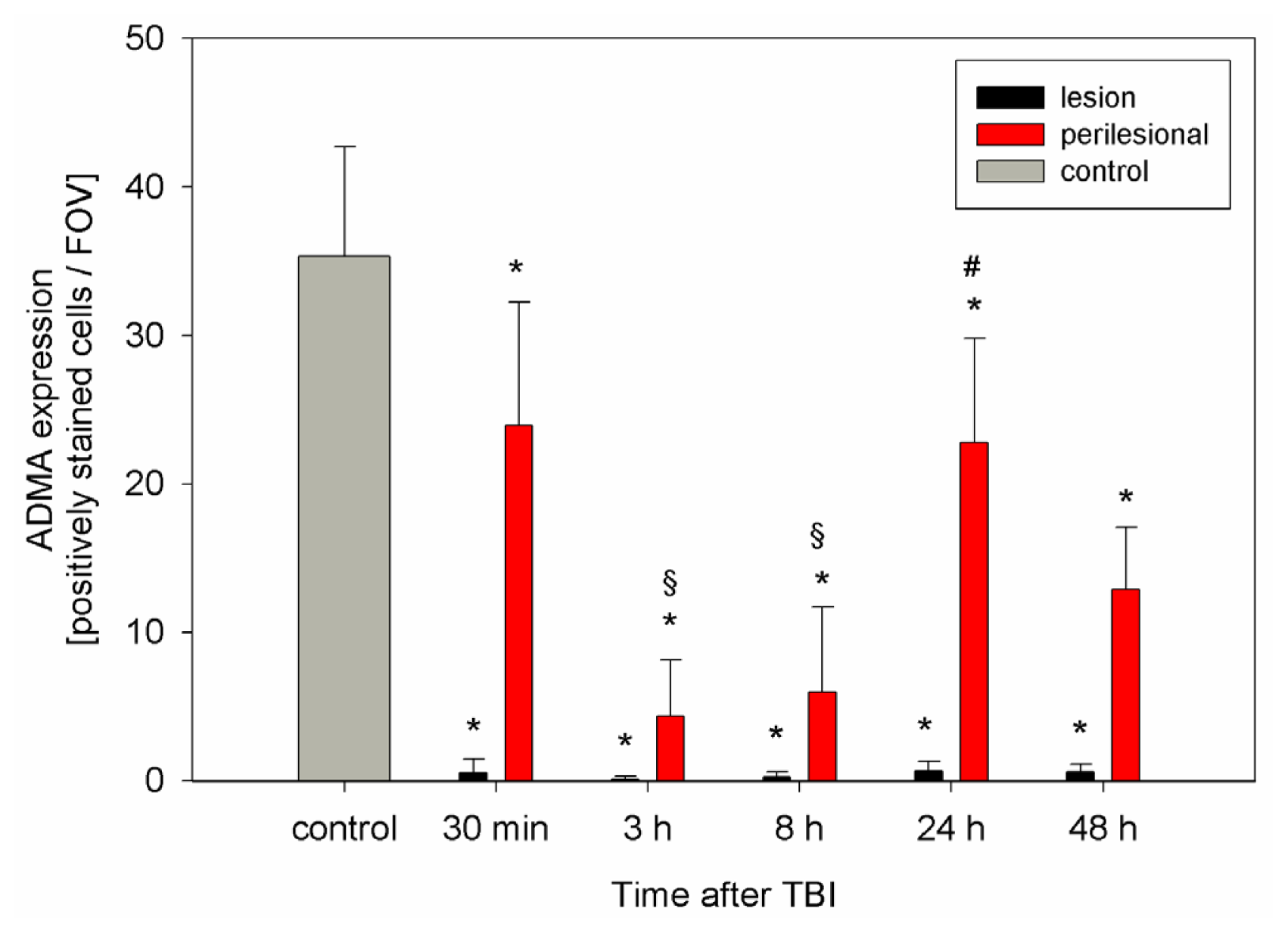
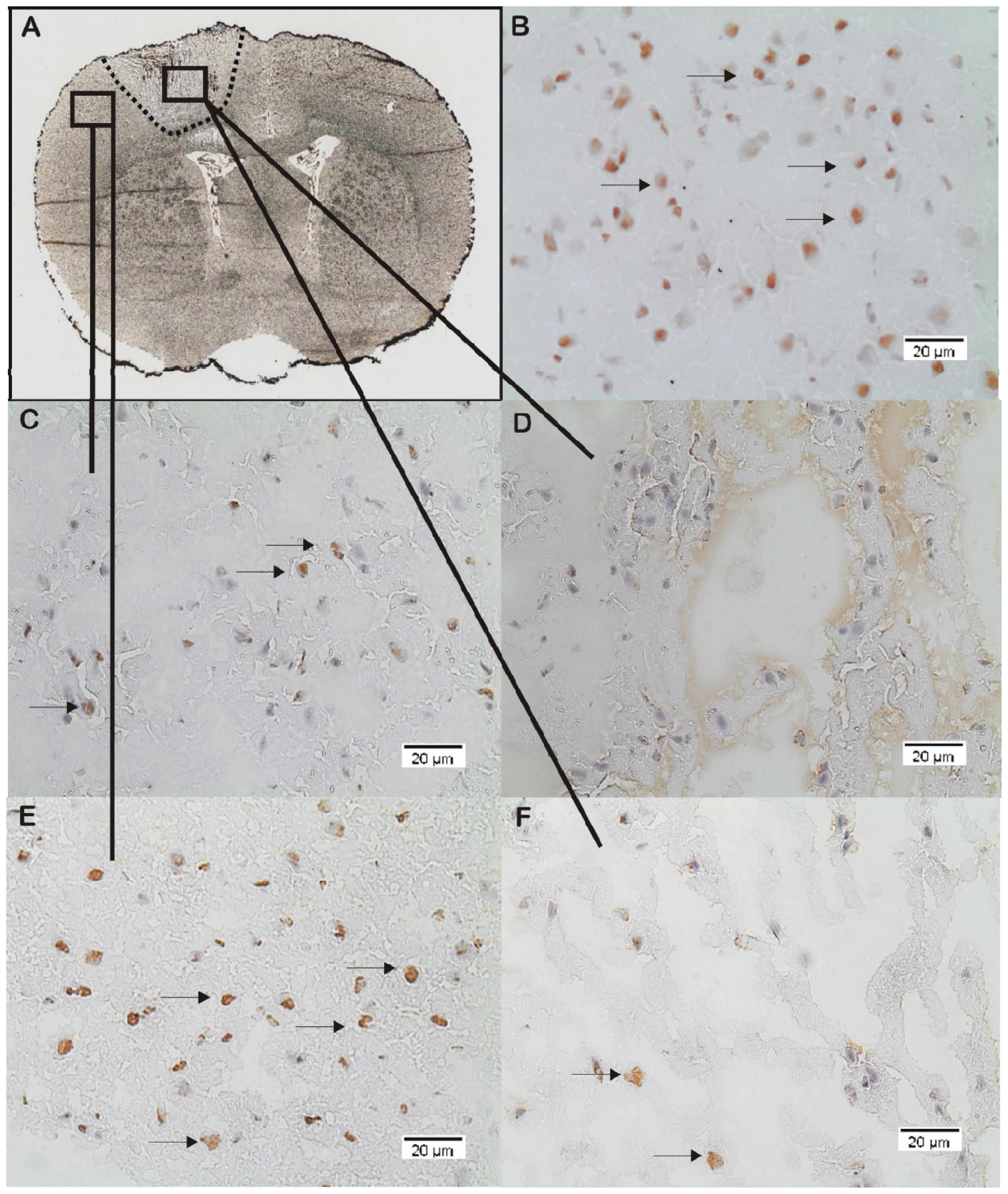
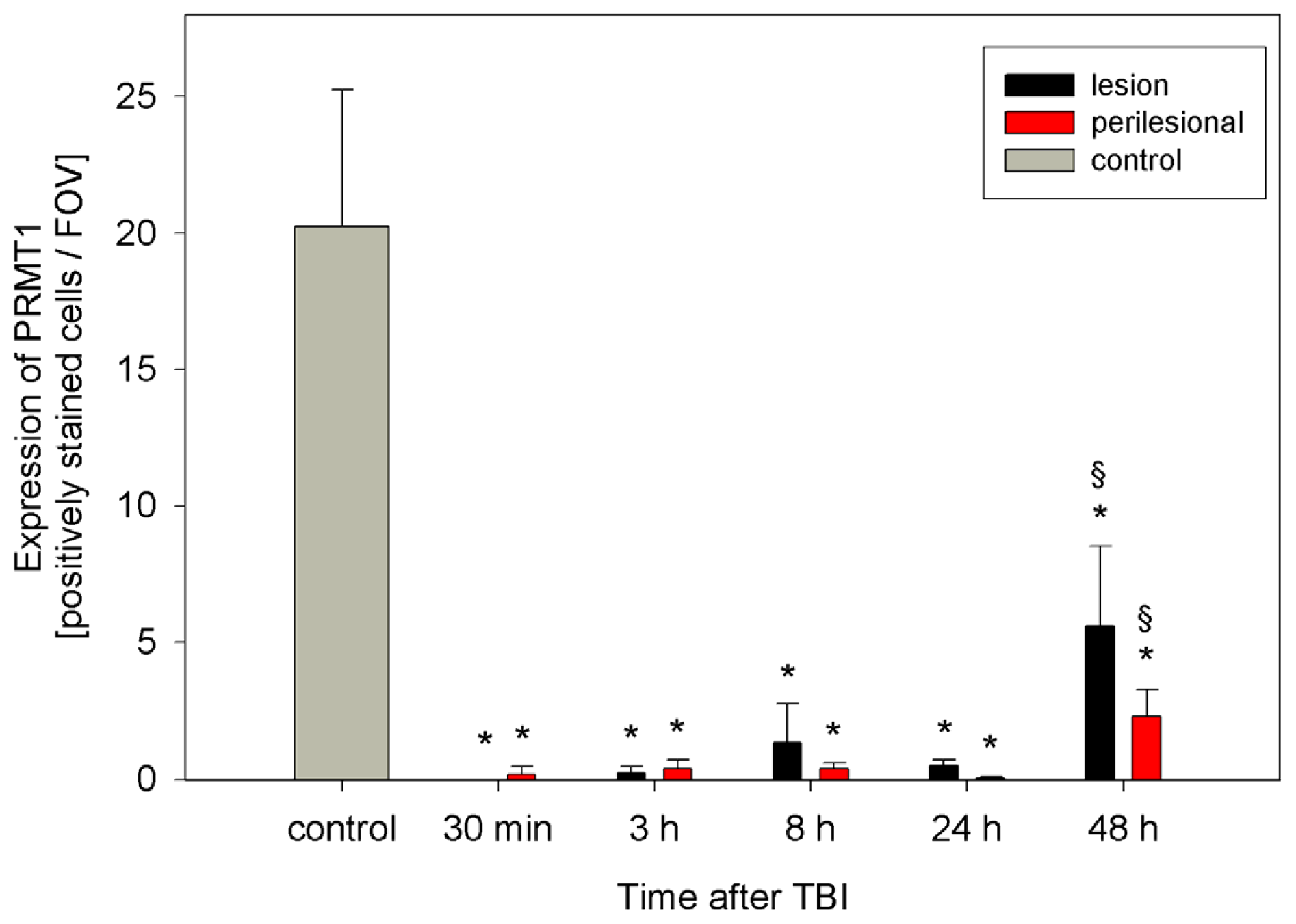
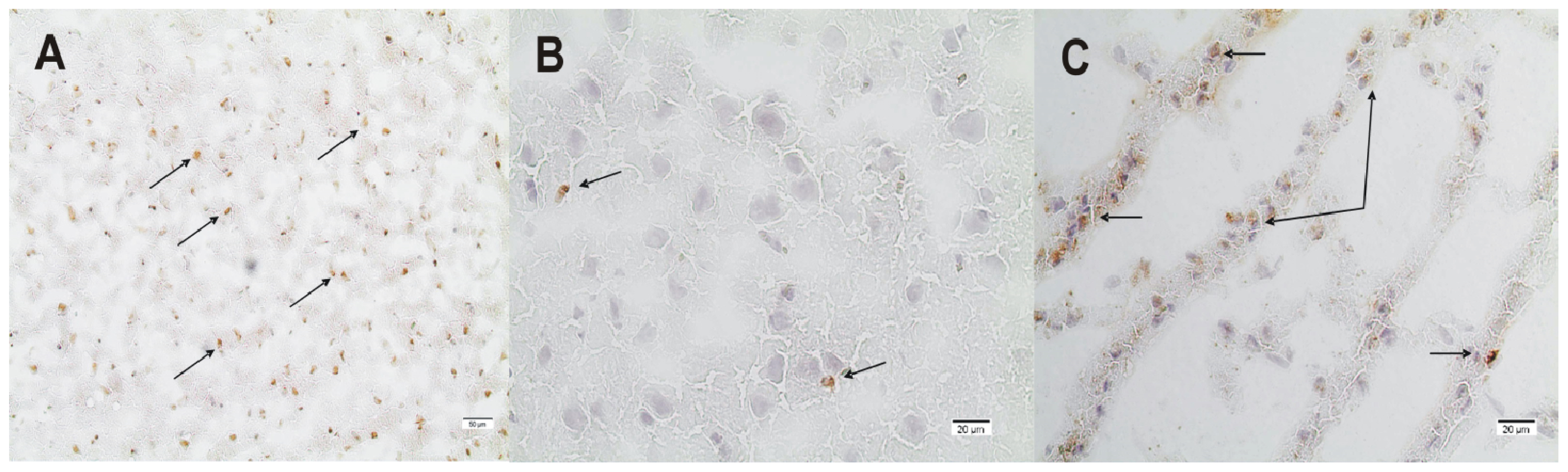
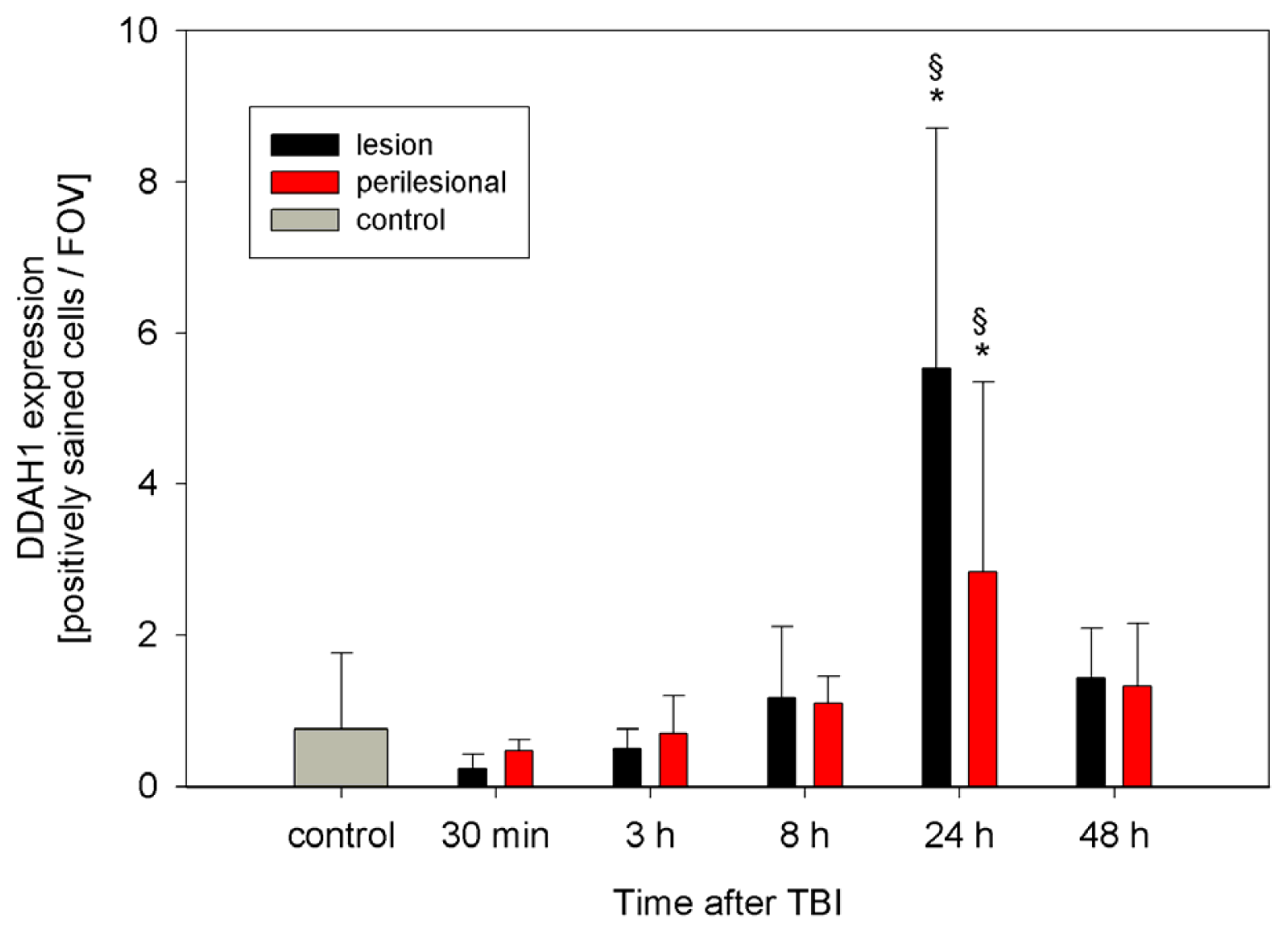
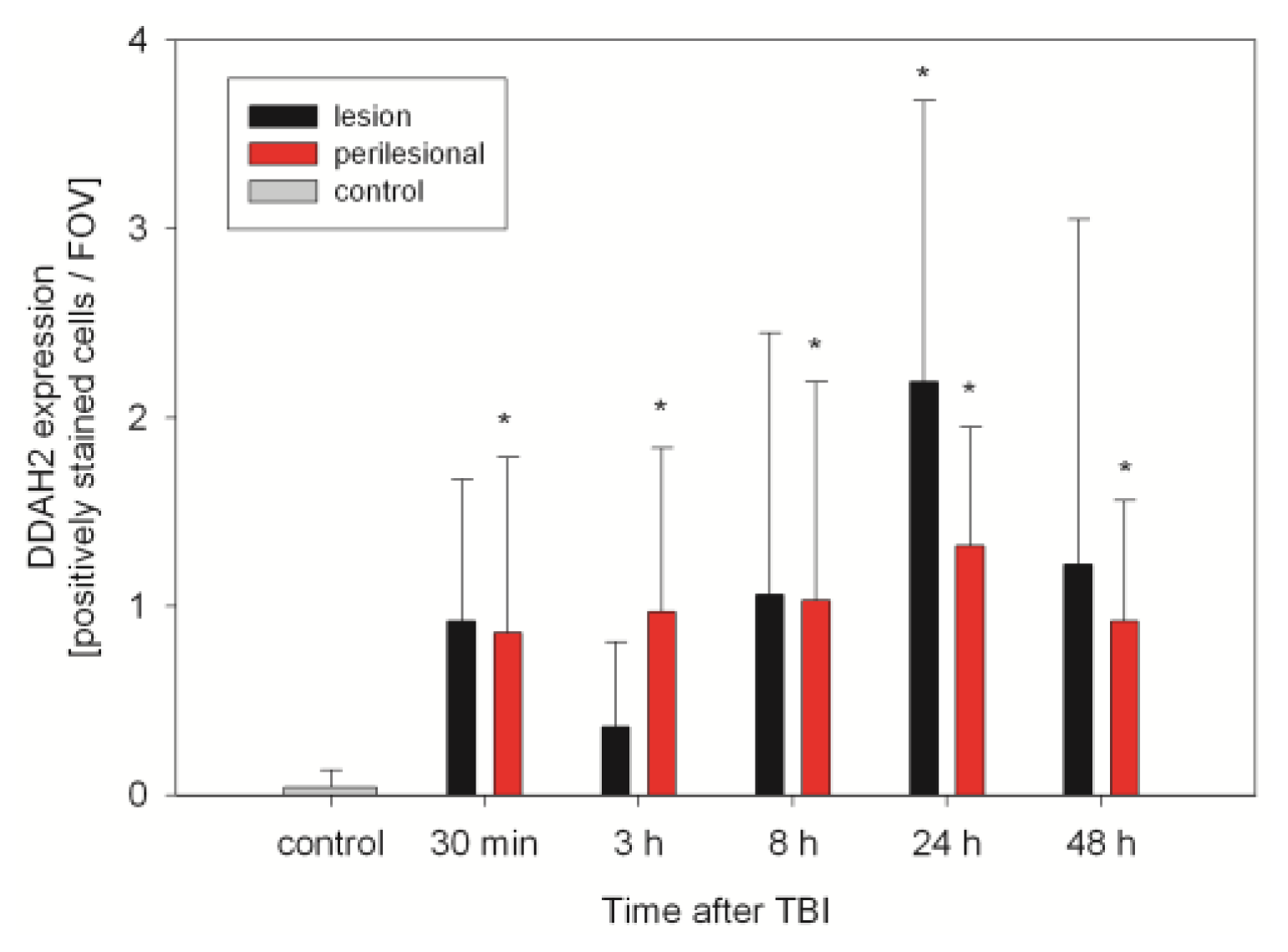
2.2. ADMA, PRMT, DDAH and NOS Expression
3. Experimental Section
3.1. Animals
3.2. Anesthesia and Brain Injury Model
3.3. Experimental Groups
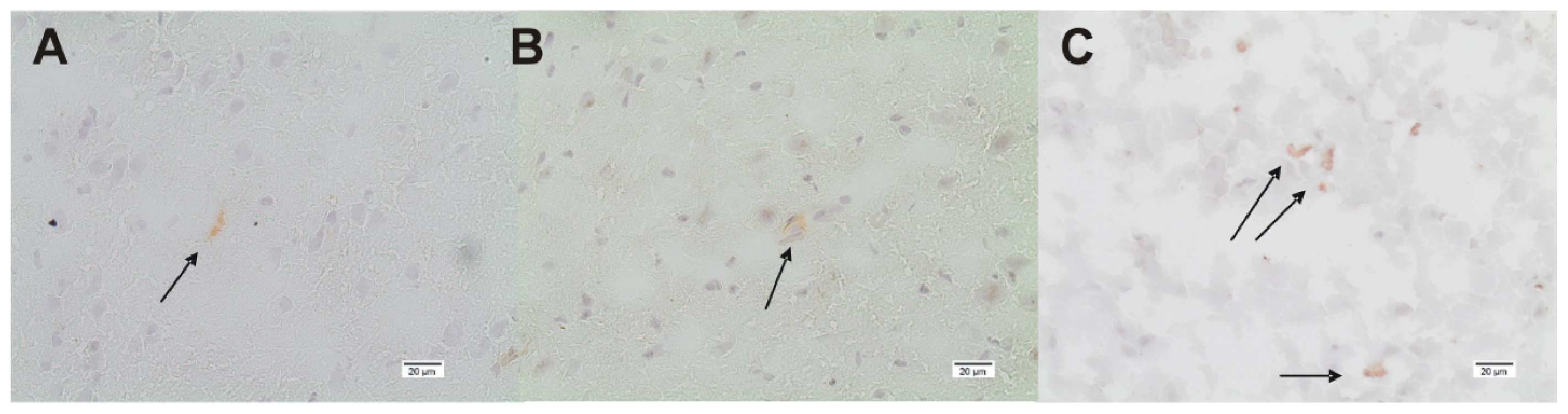
3.4. Immunohistochemistry
3.5. Neurological Assessment
3.6. Statistics
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar]
- Cherian, L.; Hlatky, R.; Robertson, C.S. Nitric oxide in traumatic brain injury. Brain Pathol. 2004, 14, 195–201. [Google Scholar]
- Pluta, R.M.; Oldfield, E.H. Analysis of nitric oxide (no) in cerebral vasospasm after aneursymal bleeding. Rev. Recent Clin. Trials 2007, 2, 59–67. [Google Scholar]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298, 249–258. [Google Scholar]
- Leiper, J.; Vallance, P. Biological significance of endogenous methylarginines that inhibit nitric oxide synthases. Cardiovasc. Res. 1999, 43, 542–548. [Google Scholar]
- Vallance, P.; Leone, A.; Calver, A.; Collier, J.; Moncada, S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar]
- Gahm, C.; Holmin, S.; Mathiesen, T. Nitric oxide synthase expression after human brain contusion. Neurosurgery 2002, 50, 1319–1326. [Google Scholar]
- Lu, Y.C.; Liu, S.; Gong, Q.Z.; Hamm, R.J.; Lyeth, B.G. Inhibition of nitric oxide synthase potentiates hypertension and increases mortality in traumatically brain-injured rats. Mol. Chem. Neuropathol. 1997, 30, 125–137. [Google Scholar]
- Mesenge, C.; Verrecchia, C.; Allix, M.; Boulu, R.R.; Plotkine, M. Reduction of the neurological deficit in mice with traumatic brain injury by nitric oxide synthase inhibitors. J. Neurotrauma 1996, 13, 209–214. [Google Scholar]
- Stoffel, M.; Rinecker, M.; Plesnila, N.; Eriskat, J.; Baethmann, A. Role of nitric oxide in the secondary expansion of a cortical brain lesion from cold injury. J. Neurotrauma 2001, 18, 425–434. [Google Scholar]
- Cherian, L.; Chacko, G.; Goodman, C.; Robertson, C.S. Neuroprotective effects of l-arginine administration after cortical impact injury in rats: Dose response and time window. J. Pharmacol. Exp. Ther. 2003, 304, 617–623. [Google Scholar]
- Cherian, L.; Chacko, G.; Goodman, J.C.; Robertson, C.S. Cerebral hemodynamic effects of phenylephrine and l-arginine after cortical impact injury. Crit. Care Med. 1999, 27, 2512–2517. [Google Scholar]
- DeWitt, D.S.; Smith, T.G.; Deyo, D.J.; Miller, K.R.; Uchida, T.; Prough, D.S. l-arginine and superoxide dismutase prevent or reverse cerebral hypoperfusion after fluid-percussion traumatic brain injury. J. Neurotrauma 1997, 14, 223–233. [Google Scholar]
- Hlatky, R.; Lui, H.; Cherian, L.; Goodman, J.C.; O’Brien, W.E.; Contant, C.F.; Robertson, C.S. The role of endothelial nitric oxide synthase in the cerebral hemodynamics after controlled cortical impact injury in mice. J. Neurotrauma 2003, 20, 995–1006. [Google Scholar]
- Wada, K.; Chatzipanteli, K.; Busto, R.; Dietrich, W.D. Role of nitric oxide in traumatic brain injury in the rat. J. Neurosurg. 1998, 89, 807–818. [Google Scholar]
- Martens-Lobenhoffer, J.; Sulyok, E.; Czeiter, E.; Buki, A.; Kohl, J.; Firsching, R.; Troger, U.; Bode-Boger, S.M. Determination of cerebrospinal fluid concentrations of arginine and dimethylarginines in patients with subarachnoid haemorrhage. J. Neurosci. Methods 2007, 164, 155–160. [Google Scholar]
- Shlosberg, D.; Benifla, M.; Kaufer, D.; Friedman, A. Blood-brain barrier breakdown as a therapeautic target in traumatic brain injury. Nat. Rev. Neurol. 2010, 6, 393–403. [Google Scholar]
- Henninger, N.; Sicard, K.M.; Li, Z.; Kulkarni, P.; Dutzmann, S.; Urbanek, C.; Schwab, S.; Fisher, M. Differential recovery of behavioral status and brain function assessed with functional magnetic resonance imaging after mild traumatic brain injury in the rat. Crit. Care Med. 2007, 35, 2607–2614. [Google Scholar]
- Kline, A.E.; Massucci, J.L.; Zafonte, R.D.; Dixon, C.E.; DeFeo, J.R.; Rogers, E.H. Differential effects of single versus multiple administrations of haloperidol and risperidone on functional outcome after experimental brain trauma. Crit. Care Med. 2007, 35, 919–924. [Google Scholar]
- Maegele, M.; Lippert-Gruener, M.; Ester-Bode, T.; Sauerland, S.; Schafer, U.; Molcanyi, M.; Lefering, R.; Bouillon, B.; Neiss, W.F.; Angelov, D.N.; et al. Reversal of neuromotor and cognitive dysfunction in an enriched environment combined with multimodal early onset stimulation after traumatic brain injury in rats. J. Neurotrauma 2005, 22, 772–782. [Google Scholar]
- Garcia, J.H.; Wagner, S.; Liu, K.F.; Hu, X.J. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats Statistical validation. Stroke 1995, 26, 627–634; discussion 635. [Google Scholar]
- Tang, J.; Frankel, A.; Cook, R.J.; Kim, S.; Paik, W.K.; Williams, K.R.; Clarke, S.; Herschman, H.R. Prmt1 is the predominant type i protein arginine methyltransferase in mammalian cells. J. Biol. Chem. 2000, 275, 7723–7730. [Google Scholar]
- Gahm, C.; Holmin, S.; Mathiesen, T. Temporal profiles and cellular sources of three nitric oxide synthase isoforms in the brain after experimental contusion. Neurosurgery 2000, 46, 169–177. [Google Scholar]
- Wada, K.; Chatzipanteli, K.; Kraydieh, S.; Busto, R.; Dietrich, W.D. Inducible nitric oxide synthase expression after traumatic brain injury and neuroprotection with aminoguanidine treatment in rats. Neurosurgery 1998, 43, 1427–1436. [Google Scholar]
- Clark, R.S.; Kochanek, P.M.; Schwarz, M.A.; Schiding, J.K.; Turner, D.S.; Chen, M.; Carlos, T.M.; Watkins, S.C. Inducible nitric oxide synthase expression in cerebrovascular smooth muscle and neutrophils after traumatic brain injury in immature rats. Pediatr. Res. 1996, 39, 784–790. [Google Scholar]
- Akaike, T.; Maeda, H. Quantification of nitric oxide using 2-phenyl-4455- tetramethylimidazoline-1-oxyl 3-oxide (ptio). Methods Enzymol. 1996, 268, 211–221. [Google Scholar]
- Jung, C.S.; Iuliano, B.A.; Harvey-White, J.; Espey, M.G.; Oldfield, E.H.; Pluta, R.M. Association between cerebrospinal fluid levels of asymmetric dimethyl-l-arginine an endogenous inhibitor of endothelial nitric oxide synthase and cerebral vasospasm in a primate model of subarachnoid hemorrhage. J. Neurosurg. 2004, 101, 836–842. [Google Scholar]
- Jung, C.S.; Oldfield, E.H.; Harvey-White, J.; Espey, M.G.; Zimmermann, M.; Seifert, V.; Pluta, R.M. Association of an endogenous inhibitor of nitric oxide synthase with cerebral vasospasm in patients with aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2007, 107, 945–950. [Google Scholar]
- Thampatty, B.P.; Klamerus, M.M.; Oberly, P.J.; Feldman, K.L.; Bell, M.J.; Tyler-Kabara, E.C.; Adelson, P.D.; Clark, R.S.; Kochanek, P.M.; Poloyac, S.M. Hypothermia decreases cerebrospinal fluid asymmetric dimethylarginine levels in children with traumatic brain injury. Pediatr. Crit. Care Med. 2013, 14, 403–412. [Google Scholar]
- Zhang, X.; Chen, Y.; Jenkins, L.W.; Kochanek, P.M.; Clark, R.S.B. Bench-to-bedside review: Apoptosis/programmed cell death triggered by traumatic brain injury. Crit. Care 2005, 9, 66–75. [Google Scholar]
- Wada, K.; Chatzipanteli, K.; Busto, R.; Dietrich, W.D. Effects of l-name and 7-ni on nos catalytic activity and behavioral outcome after traumatic brain injury in the rat. J. Neurotrauma 1999, 16, 203–212. [Google Scholar]
- Goksel, H.M.; Ozum, U.; Oztoprak, I. The therapeutic effect of continuous intracisternal l-arginine infusion on experimental cerebral vasospasm. Acta Neurochir. (Wien.) 2001, 143, 277–285. [Google Scholar]
- Pluta, R.M.; Afshar, J.K.; Thompson, B.G.; Boock, R.J.; Harvey-White, J.; Oldfield, E.H. Increased cerebral blood flow but no reversal or prevention of vasospasm in response to l-arginine infusion after subarachnoid hemorrhage. J. Neurosurg. 2000, 92, 121–126. [Google Scholar]
- Huk, I.; Nanobashvili, J.; Neumayer, C.; Punz, A.; Mueller, M.; Afkhampour, K.; Mittlboeck, M.; Losert, U.; Polterauer, P.; Roth, E.; et al. l-arginine treatment alters the kinetics of nitric oxide and superoxide release and reduces ischemia/reperfusion injury in skeletal muscle. Circulation 1997, 96, 667–675. [Google Scholar]
- Yamaguchi, A.; Kitajo, K. The effect of prmt1-mediated arginine methylation on the subcellular localization stress granules and detergent-insoluble aggregates of fus/tls. PLoS One 2012, 7, e49267. [Google Scholar]
- Tran, C.T.; Fox, M.F.; Vallance, P.; Leiper, J.M. Chromosomal localization gene structure and expression pattern of ddah1: Comparison with ddah2 and implications for evolutionary origins. Genomics 2000, 68, 101–105. [Google Scholar]
- Ogawa, T.; Kimoto, M.; Sasaoka, K. Purification and properties of a new enzyme ngng-dimethylarginine dimethylaminohydrolase from rat kidney. J. Biol. Chem. 1989, 264, 10205–10209. [Google Scholar]
- Leiper, J.M.; Santa Maria, J.; Chubb, A.; MacAllister, R.J.; Charles, I.G.; Whitley, G.S.; Vallance, P. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem. J. 1999, 34, 209–214. [Google Scholar]
- Amrouni, D.; Meiller, A.; Gautier-Sauvigne, S.; Piraud, M.; Bouteille, B.; Vincendeau, P.; Buguet, A.; Cespuglio, R. Cerebral changes occurring in arginase and dimethylarginine dimethylaminohydrolase (ddah) in a rat model of sleeping sickness. PLoS One 2011, 6, e16891. [Google Scholar]
- Boger, R.H.; Sydow, K.; Borlak, J.; Thum, T.; Lenzen, H.; Schubert, B.; Tsikas, D.; Bode-Boger, S.M. Ldl cholesterol upregulates synthesis of asymmetrical dimethylarginine in human endothelial cells: Involvement of S-adenosylmethionine-dependent methyltransferases. Circ. Res. 2000, 87, 99–105. [Google Scholar]
- Ito, A.; Tsao, P.S.; Adimoolam, S.; Kimoto, M.; Ogawa, T.; Cooke, J.P. Novel mechanism for endothelial dysfunction: Dysregulation of dimethylarginine dimethylaminohydrolase. Circulation 1999, 99, 3092–3095. [Google Scholar]
- Immonen, R.; Heikkinen, T.; Tahtivaara, L.; Nurmi, A.; Stenius, T.K.; Puolivali, J.; Tuinstra, T.; Phinney, A.L.; van Vliet, B.; Yrjanheikki, J.; et al. Cerebral blood volume alterations in the perilesional areas in the rat brain after traumatic brain injury—Ccomparison with behavioral outcome. J. Cereb. Blood Flow Metab. 2010, 30, 1318–1328. [Google Scholar]
- Linsky, T.W.; Fast, W. Discovery of structurally-diverse inhibitor scaffolds by high-throughput screening of a fragment library with dimethylarginine dimethylaminohydrolase. Bioorg. Med. Chem. 2012, 20, 5550–5558. [Google Scholar]
- Nandi, M.; Kelly, P.; Torondel, B.; Wang, Z.; Starr, A.; Ma, Y.; Cunningham, P.; Stidwill, R.; Leiper, J. Genetic and pharmacological inhibition of dimethylarginine dimethylaminohydrolase 1 is protective in endotoxic shock. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2589–2597. [Google Scholar]
- Zweckberger, K.; Eros, C.; Zimmermann, R.; Kim, S.W.; Engel, D.; Plesnila, N. Effect of early and delayed decompressive craniectomy on secondary brain damage after controlled cortical impact in mice. J. Neurotrauma 2006, 23, 1083–1093. [Google Scholar]










© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jung, C.S.; Wispel, C.; Zweckberger, K.; Beynon, C.; Hertle, D.; Sakowitz, O.W.; Unterberg, A.W. Endogenous Nitric-Oxide Synthase Inhibitor ADMA after Acute Brain Injury. Int. J. Mol. Sci. 2014, 15, 4088-4103. https://doi.org/10.3390/ijms15034088
Jung CS, Wispel C, Zweckberger K, Beynon C, Hertle D, Sakowitz OW, Unterberg AW. Endogenous Nitric-Oxide Synthase Inhibitor ADMA after Acute Brain Injury. International Journal of Molecular Sciences. 2014; 15(3):4088-4103. https://doi.org/10.3390/ijms15034088
Chicago/Turabian StyleJung, Carla S., Christian Wispel, Klaus Zweckberger, Christopher Beynon, Daniel Hertle, Oliver W. Sakowitz, and Andreas W. Unterberg. 2014. "Endogenous Nitric-Oxide Synthase Inhibitor ADMA after Acute Brain Injury" International Journal of Molecular Sciences 15, no. 3: 4088-4103. https://doi.org/10.3390/ijms15034088
APA StyleJung, C. S., Wispel, C., Zweckberger, K., Beynon, C., Hertle, D., Sakowitz, O. W., & Unterberg, A. W. (2014). Endogenous Nitric-Oxide Synthase Inhibitor ADMA after Acute Brain Injury. International Journal of Molecular Sciences, 15(3), 4088-4103. https://doi.org/10.3390/ijms15034088