Exogenous Asymmetric Dimethylarginine (ADMA) in Pathogenesis of Ischemia-Reperfusion-Induced Gastric Lesions: Interaction with Protective Nitric Oxide (NO) and Calcitonin Gene-Related Peptide (CGRP)

 ,
, 

Abstract
:1. Introduction
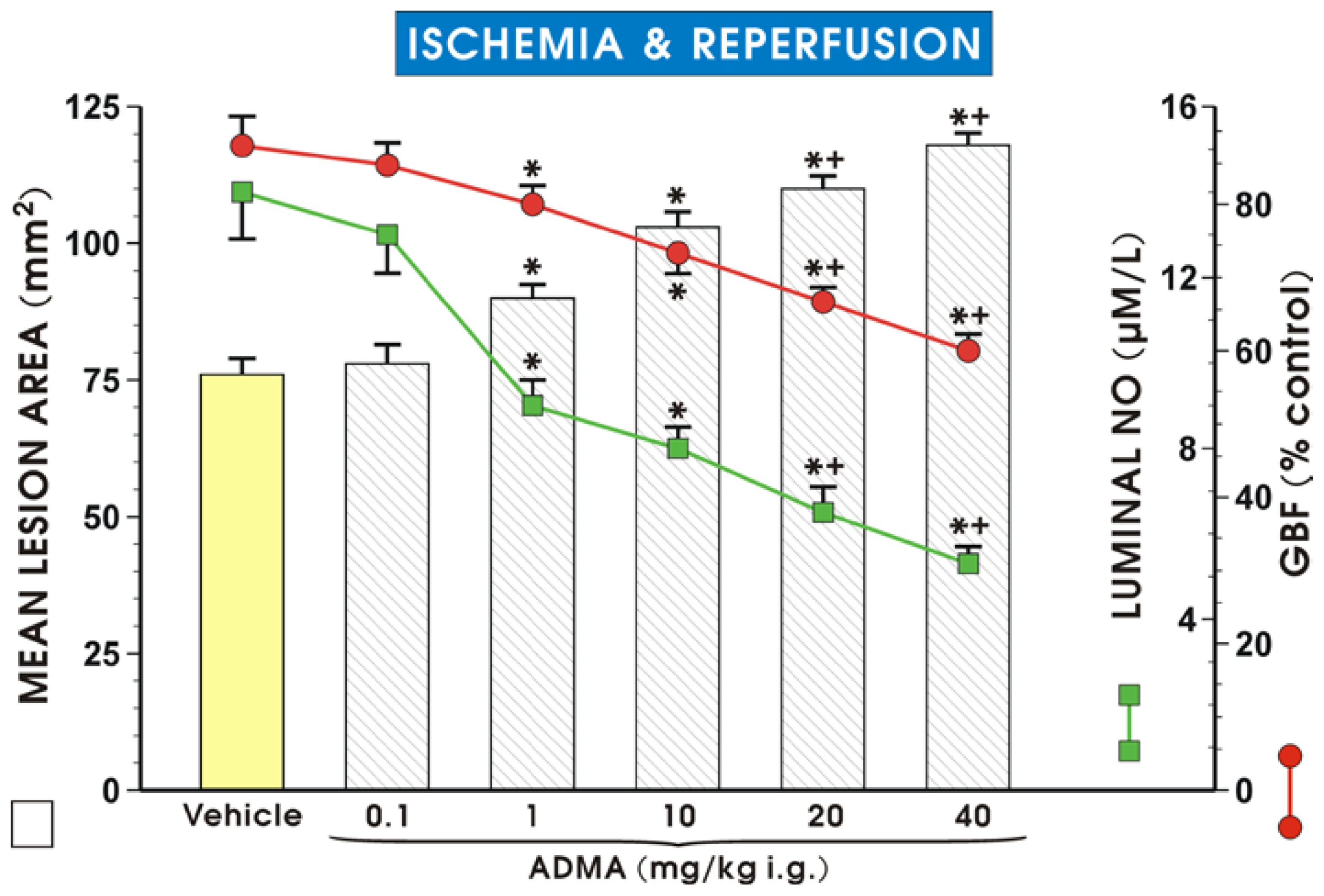
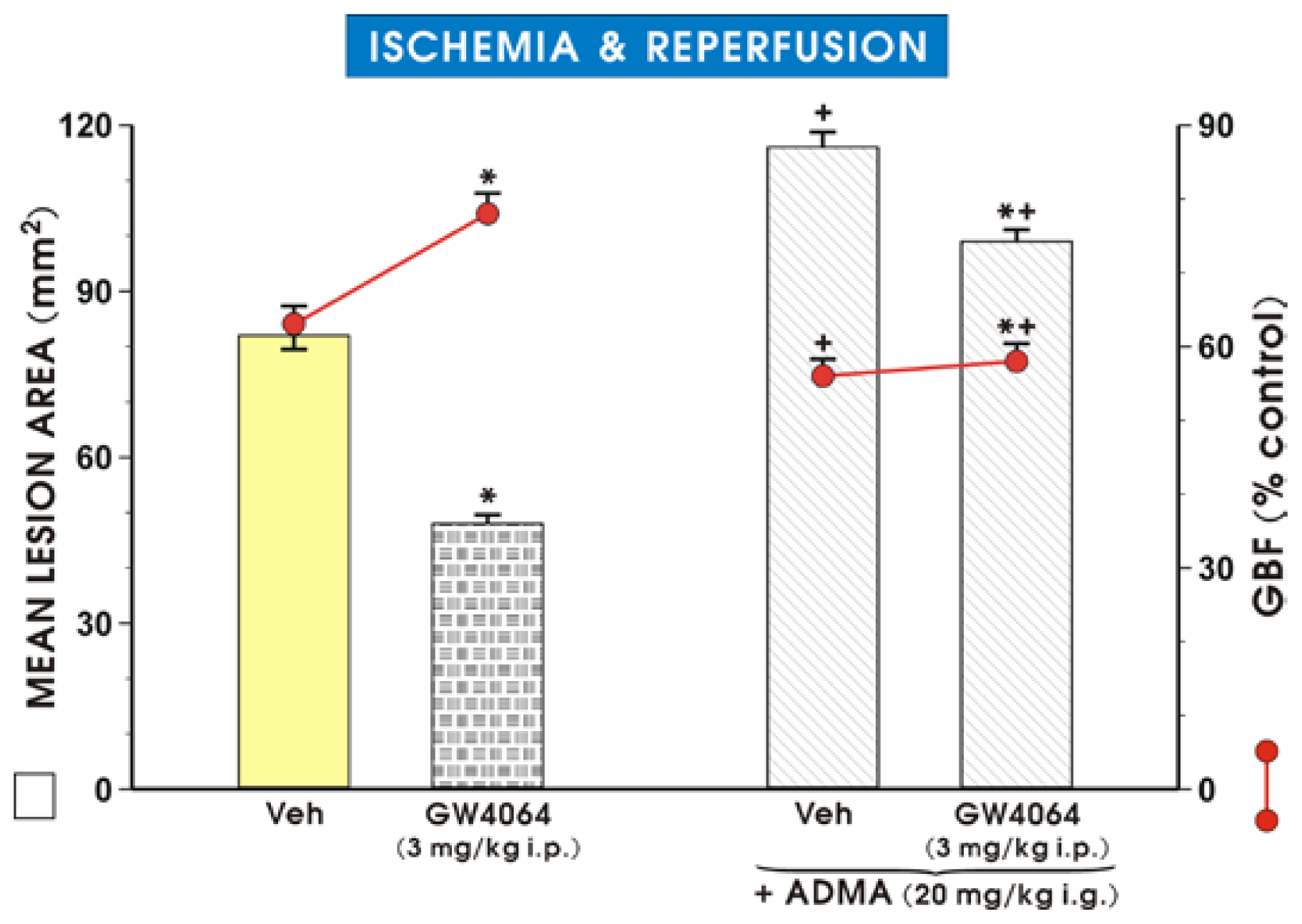
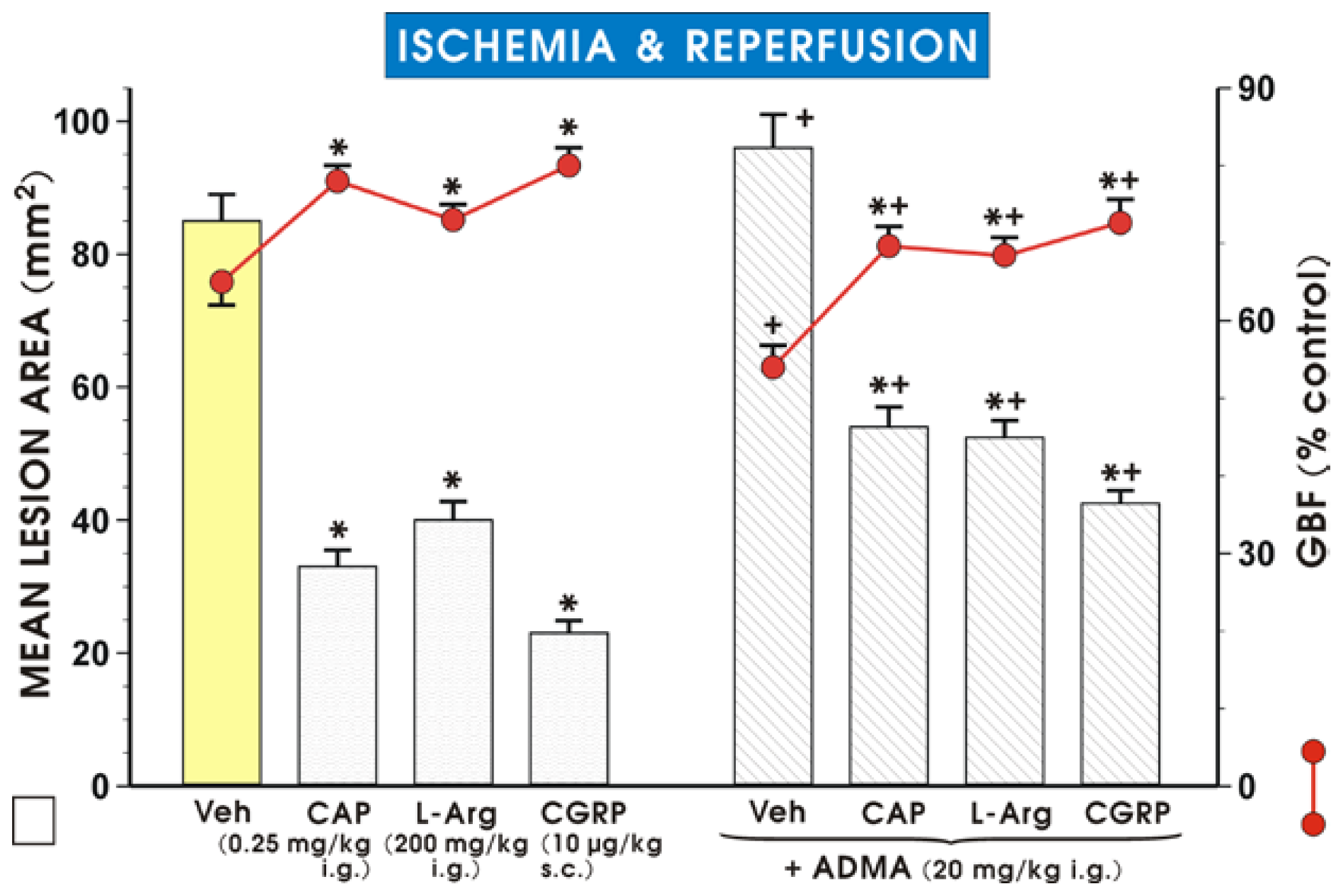
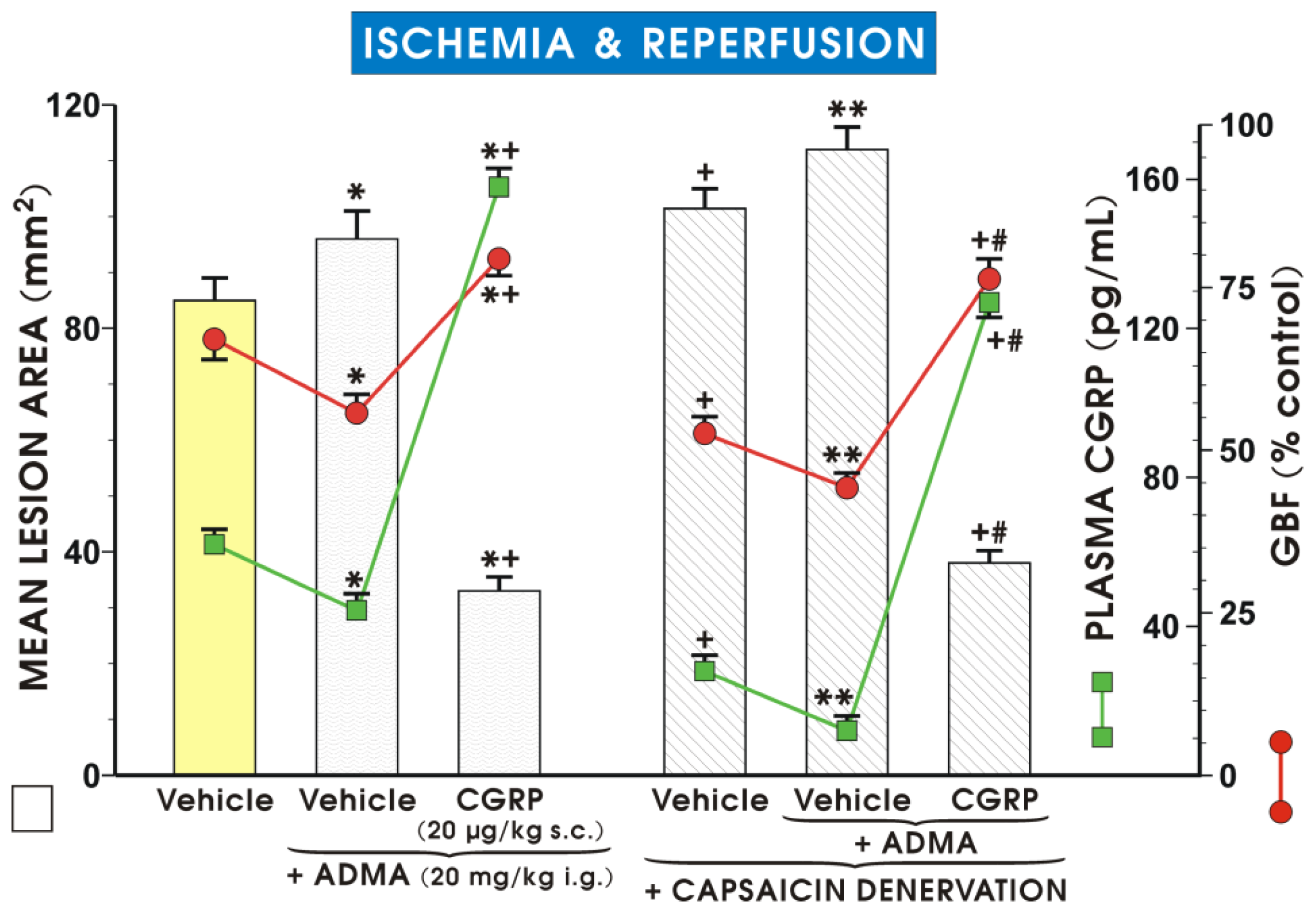
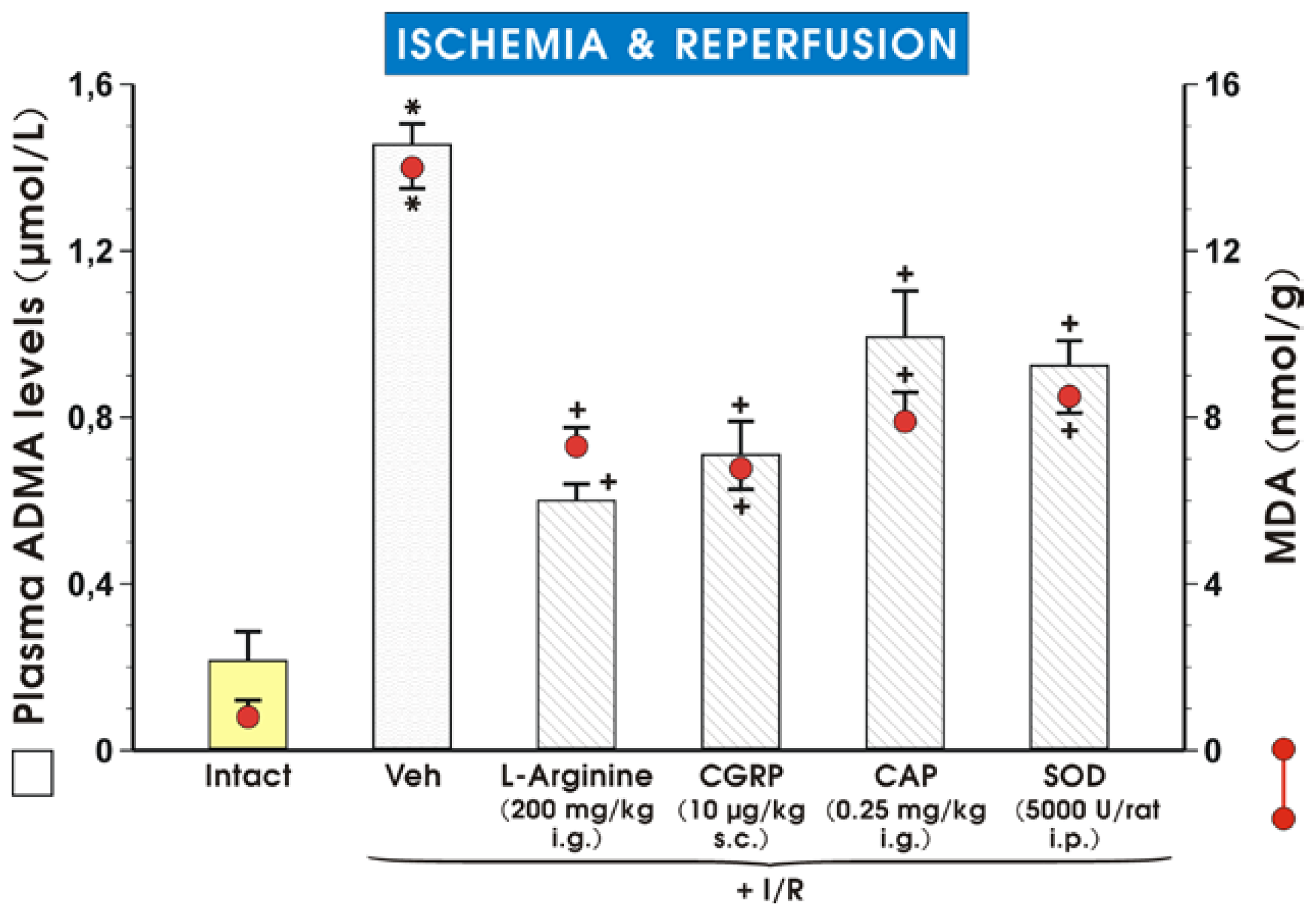
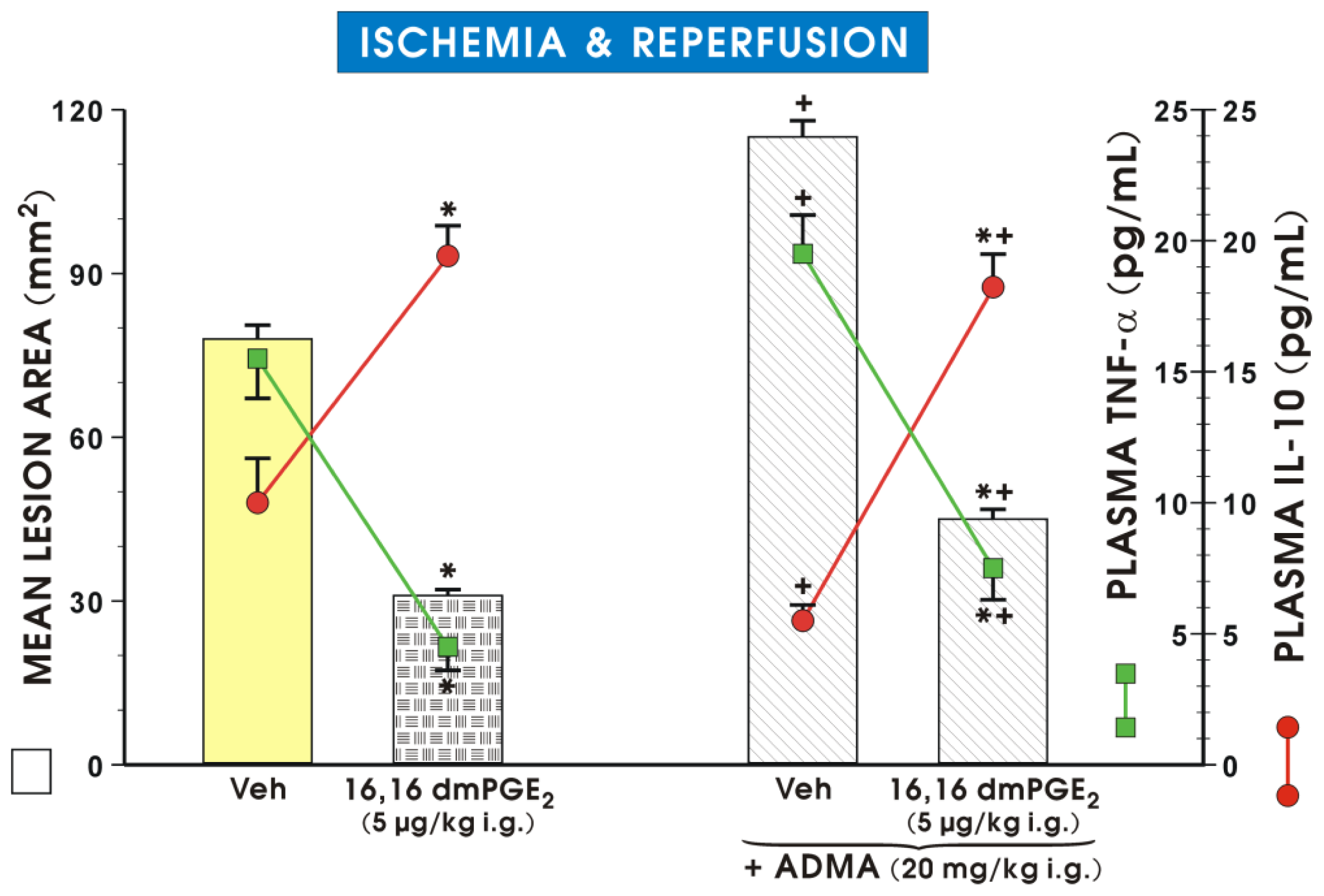
2. Results and Discussion
3. Experimental Section
3.1. Animals, Ischemia/Reperfusion Injury, Afferent Sensory Nerves Ablation
3.2. Determination of Gastric Blood Flow and the Area of Gastric Lesions
3.3. Determination of Luminal NO Release and the Gastric Mucosal MDA Content
3.4. Assessment of Plasma Levels of ADMA, CGRP and Proinflammatory and Anti-Inflammatory Cytokines
3.5. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Zakrzewicz, D.; Eickelberg, O. From arginine methylation to ADMA: A novel mechanism with therapeutic potential in chronic lung diseases. BCM Pulm. Med. 2009, 9, 5. [Google Scholar]
- Leiper, J.; Vallance, P. Biological significance of endogenous methylarginines that inhibit nitric oxide synthases. Cardiovasc Res. 1999, 43, 542–548. [Google Scholar]
- Leiper, J.; Vallance, P. The synthesis and metabolism of asymmetric dimethylarginine (ADMA). Eur. J. Clin. Pharmacol. 2006, 62, 33–38. [Google Scholar]
- Beltowski, J.; Kedra, A. Asymmetric dimethylarginine (ADMA) as a target for pharmacotherapy. Pharmacol. Rep. 2006, 58, 159–178. [Google Scholar]
- Wilcken, D.E.; Sim, A.S.; Wang, J.; Wang, X.L. Asymmetric dimethylarginine (ADMA) in vascular renal and hepatic disease and the regulatory role of l-arginine on its metabolism. Mol. Genet. Metab. 2007, 91, 309–317. [Google Scholar]
- Vallance, P.; Leiper, J. Cardiovascular biology of the asymmetric dimethylarginine dimethylaminohydrolase pathway. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1023–1030. [Google Scholar]
- Cardounel, A.J.; Cui, H.; Samouilov, A.; Johnson, W.; Kearns, P.; Tsai, A.L.; Berka, V.; Zweier, J.L. Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular function. J. Biol. Chem. 2007, 282, 879–887. [Google Scholar]
- Szlachcic, A.; Krzysiek-Maczka, G.; Pajdo, R.; Targosz, A.; Magierowski, M.; Jasnos, K.; Drozdowicz, D.; Kwiecien, S.; Brzozowski, T. The impact of asymmetric dimethylarginine (ADMA) the endogenous nitric oxide (NO) synthase inhibitor to the pathogenesis of gastric mucosal damage. Curr. Pharm. Des. 2013, 19, 90–97. [Google Scholar]
- Zhang, G.G.; Bai, Y.P.; Chen, M.F.; Shi, R.Z.; Jiang, D.J.; Fu, Q.M.; Tan, G.S.; Li, Y.J. Asymmetric dimethylarginine induces TNF-alpha production via ROS/NF-kappaB dependent pathway in human monocytic cells and the inhibitory effect of retinioside C. Vasc. Pharmacol. 2008, 48, 115–121. [Google Scholar]
- Chen, M.; Li, Y.; Yang, T.; Wang, Y.; Bai, Y.; Xie, X. ADMA induces monocyte adhesion via activation of chemokine receptors in cultured THP-1 cells. Cytokine 2008, 43, 149–159. [Google Scholar]
- Landim, M.B.; Casella Filho, A.; Chagas, A.C. Asymmetric dimethylarginine (ADMA) and endothelial dysfunction: implications for atherogenesis. Clinics 2009, 64, 471–478. [Google Scholar]
- Zhang, Z.; Zhou, Y.; Zou, Y.Y.; Wang, L.; Yang, Z.C.; Guo, R.; Li, D.; Peng, J.; Li, Y.J. Detrimental effects of nicotine on the acute gastric mucosal injury induced by ethanol: Role of asymmetric dimethylarginine. Can. J. Physiol. Pharmacol. 2008, 86, 835–840. [Google Scholar]
- Kwiecien, S.; Ptak-Belowska, A.; Krzysiek-Maczka, G.; Targosz, A.; Jasnos, K.; Magierowski, M.; Szczyrk, U.; Brzozowski, B.; Konturek, S.J.; Konturek, P.C.; et al. Asymmetric dimethylarginine an endogenous inhibitor of nitric oxide synthase interacts with gastric oxidative metabolism and enhances stress-induced gastric lesions. J. Physiol. Pharmacol. 2012, 63, 515–524. [Google Scholar]
- Ogawa, T.; Kimoto, M.; Sasaoka, K. Occurrence of a new enzyme catalyzing the direct conversion of NG NG-dimethyl-l-arginine to l-citrulline in rats. Biochem. Biophys. Res. Commun. 1987, 148, 671–677. [Google Scholar]
- Liu, X.; Fassett, J.; Wei, Y.; Chen, Y. Regulation of DDAH1 as a potential therapeutic target for treating cardiovascular diseases. Evid. Based Complement. Alternat. Med. 2013. [Google Scholar] [CrossRef]
- Vaquero, J.; Briz, O.; Herraez, E.; Muntané, J.; Marin, J.J. Activation of the nuclear receptor FXR enhances hepatocyte chemoprotection and liver tumor chemoresistance against genotoxic compounds. Biochim. Biophys. Acta 2013, 1833, 2212–2219. [Google Scholar]
- Li, J.; Wilson, A.; Gao, X.; Kuruba, R.; Liu, Y.; Poloyac, S.; Pitt, B.; Xie, W.; Li, S. Coordinated regulation of dimethylarginine dimethylaminohydrolase-1 and cationic amino acid transporter-1 by farnesoid X receptor in mouse liver and kidney and its implication in the control of blood levels of asymmetric dimethylarginine. J. Pharmacol. Exp. Ther 2009, 331, 234–243. [Google Scholar]
- Brzozowski, T.; Konturek, S.J.; Sliwowski, Z.; Pytko-Polonczyk, J.; Szlachcic, A.; Drozdowicz, D. Role of capsaicin-sensitive sensory nerves in gastroprotection against acid-independent and acid-dependent ulcerogens. Digestion 1996, 57, 424–432. [Google Scholar]
- Villegas, I.; Martin, A.R.; Toma, W.; de la Lastra, C.A. Rosiglitazone an agonist of peroxisome proliferator-activated receptor gamma protects against gastric ischemia-reperfusion damage in rats: Role of oxygen free radicals generation. Eur. J. Pharmacol. 2004, 505, 195–203. [Google Scholar]
- Flögel, U.; Decking, U.K.; Gödecke, A.; Schrader, J. Contribution of NO to ischemia-reperfusion injury in the saline-perfused heart: A study in endothelial NO synthase knockout mice. J. Mol. Cell. Cardiol. 1999, 31, 827–836. [Google Scholar]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar]
- Chatterjee, P.K.; Patel, N.S.; Kvale, E.O.; Cuzzocrea, S.; Brown, P.A.; Stewart, K.N.; Mota-Filipe, H.; Thiemmermann, C. Inhibition of inducible nitric oxide synthase reduces renal ischemia/reperfusion injury. Kidney Int. 2002, 61, 862–871. [Google Scholar]
- Andelová, E.; Barteková, M.; Pancza, D.; Styk, J.; Ravingerová, T. The role of NO in ischemia/reperfusion injury in isolated rat heart. Gen. Physiol. Biophys. 2005, 2, 411–426. [Google Scholar]
- Phan, L.H.; Hickey, M.J.; Niazi, Z.B.; Stewart, A.G. Nitric oxide synthase inhibitor nitro-iminoethyl-l-ornithine reduces ischemia-reperfusion injury in rabbit skeletal muscle. Microsurgery 1994, 15, 703–707. [Google Scholar]
- Andrews, F.J.; Malcontenti-Wilson, C.; O’Brien, P.E. Protection against gastric ischemiareperfusion injury by nitric oxide generators. Dig. Dis. Sci. 1994, 39, 366–373. [Google Scholar]
- Wang, L.; Zhou, Y.; Peng, J.; Zhang, Z.; Jiang, D.J.; Li, Y.J. Role of endogenous nitric oxide synthase inhibitor in gastric mucosal injury. Can. J. Physiol. Pharmacol. 2008, 86, 97–104. [Google Scholar]
- Li, L.; Luo, X.J.; Liu, Y.Z.; Zhang, Y.S.; Yuan, Q.; Tan, N.; Xiang, D.X.; Peng, J. The role of the DDAH-ADMA pathway in the protective effect of resveratrol analog BTM-0512 on gastric mucosal injury. Can. J. Physiol. Pharmacol. 2010, 88, 562–567. [Google Scholar]
- Zhang, Z.; Zou, Y.Y.; Li, F.J.; Hu, C.P. Asymmetric dimethylarginine: A novel biomarker of gastric mucosal injury? World J. Gastroenterol. 2011, 17, 2178–2180. [Google Scholar]
- Robert, A. Cytoprotection of the gastrointestinal mucosa. Adv. Intern. Med 1983, 28, 325–337. [Google Scholar]
- Brzozowski, T.; Konturek, P.C.; Konturek, S.J.; Brzozowska, I.; Pawlik, T. Role of prostaglandins in gastroprotection and gastric adaptation. J Physiol Pharmacol 2005, 56, 33–55. [Google Scholar]
- Stühlinger, M.C.; Conci, E.; Haubner, B.J.; Stocker, E.M.; Schwaighofer, J.; Cooke, J.P.; Tsao, P.S.; Pachinger, O.; Metzler, B. Asymmetric dimethyl l-arginine (ADMA) is a critical regulator of myocardial reperfusion injury. Cardiovasc. Res. 2007, 75, 417–425. [Google Scholar]
- Kim, H.; Hwan Kim, K. Role of nitric oxide and mucus in ischemia/reperfusion-induced gastric mucosal injury in rats. Pharmacology 2001, 62, 200–207. [Google Scholar]
- Kobata, A.; Kotani, T.; Komatsu, Y.; Amagase, K.; Kato, S.; Takeuchi, K. Dual action of nitric oxide in the pathogenesis of ischemia/reperfusion-induced mucosal injury in mouse stomach. Digestion 2007, 75, 188–197. [Google Scholar]
- Boger, R.H. Asymmetric dimethylarginine an endogenous inhibitor of nitric oxide synthase explains the “l-arginine paradox” and acts as a novel cardiovascular risk factor. J Nutr 2004, 134, 2842–2847. [Google Scholar]
- Berg, A.; Redéen, S.; Grenegård, M.; Ericson, A.C.; Sjöstrand, S.E. Nitric oxide inhibits gastric acid secretion by increasing intraparietal cell levels of cGMP in isolated human gastric glands. Am. J. Physiol. Gastrointest. Liver Physiol 2005, 289, 1061–1066. [Google Scholar]
- Combet, E.; Preston, T.; McColl, K.E. Development of an in vitro system combining aqueous and lipid phases as a tool to understand gastric nitrosation. Rapid Commun. Mass Spectrom 2010, 24, 529–534. [Google Scholar]
- Shibata, N.; Matsui, H.; Yokota, T.; Matsuura, B.; Maeyama, K.; Onji, M. Direct effects of nitric oxide on histamine release from rat enterochromaffin-like cells. Eur. J. Pharmacol 2006, 535, 25–33. [Google Scholar]
- Brzozowski, T.; Konturek, P.C.; Konturek, S.J.; Drozdowicz, D.; Kwiecien, S.; Pajdo, R.; Bielanski, W.; Hahn, E.G. Role of gastric acid secretion in progression of acute gastric erosions induced by ischemia-reperfusion into gastric ulcers. Eur. J. Pharmacol. 2000, 398, 147–158. [Google Scholar]
- Rocha, B.S.; Gago, B.; Barbosa, R.M.; Lundberg, J.O.; Radi, R.; Laranjinha, J. Intragastric nitration by dietary nitrite: implications for modulation of protein and lipid signaling. Free Radic. Biol. Med 2012, 52, 693–698. [Google Scholar]
- Brzozowski, T.; Konturek, P.C.; Konturek, S.J.; Pajdo, R.; Kwiecien, S.; Pawlik, M.; Drozdowicz, D.; Sliwowski, Z.; Pawlik, W.W. Ischemic preconditioning of remote organs attenuates gastric ischemia-reperfusion injury through involvement of prostaglandins and sensory nerves. Eur. J. Pharmacol. 2004, 499, 201–213. [Google Scholar]
- Kwiecien, S.; Pawlik, M.W.; Sliwowski, Z.; Kwiecien, N.; Brzozowski, T.; Pawlik, W.W.; Konturek, S.J. Involvement of sensory afferent fibers and lipid peroxidation in the pathogenesis of stress-induced gastric mucosa damage. J. Physiol. Pharmacol. 2007, 58, 149–162. [Google Scholar]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev 2009, 240–273. [Google Scholar]
- Elinav, E.; Henao-Mejia, J.; Flavell, R.A. Integrative inflammasome activity in the regulation of intestinal mucosal immune responses. Mucosal Immunol 2013, 6, 4–13. [Google Scholar]
- Sandanger, ⊘; Ranheim, T.; Vinge, L.E.; Bliksøen, M.; Alfsnes, K.; Finsen, A.V.; Dahl, C.P.; Askevold, E.T.; Florholmen, G.; Christensen, G.; et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischemia-reperfusion injury. Cardiovasc. Res 2013, 99, 164–174. [Google Scholar]
- Fann, D.Y.; Lee, S.Y.; Manzanero, S.; Chunduri, P.; Sobey, C.G.; Arumugam, T.V. Pathogenesis of acute stroke and the role of inflammasomes. Ageing Res. Rev 2013, 12, 941–966. [Google Scholar]
- Tarnawski, A.; Brzozowski, T.; Sarfeh, I.J.; Krause, W.J.; Ulich, T.R.; Gergely, H.; Hollander, D. Prostaglandin protection of human isolated gastric glands against indomethacin and ethanol injury Evidence for direct cellular action of prostaglandin. J. Clin. Investig 1988, 81, 1081–1089. [Google Scholar]
- Brzozowski, T.; Tarnawski, A.; Hollander, D.; Sekhon, S.; Krause, W.J.; Gergely, H. Comparison of prostaglandin and cimetidine in protection of isolated gastric glands against indomethacin injury. J. Physiol. Pharmacol 2005, 56, 75–88. [Google Scholar]
- Pajdo, R.; Brzozowski, T.; Konturek, P.C.; Kwiecien, S.; Konturek, S.J.; Sliwowski, Z.; Pawlik, M.; Ptak, A.; Drozdowicz, D.; Hahn, E.G. Ischemic preconditioning the most effective gastroprotective intervention: involvement of prostaglandins nitric oxide adenosine and sensory nerves. Eur. J. Pharmacol 2001, 427, 263–276. [Google Scholar]
- Wu, Y.H.; Zhang, X.; Wang, D.H. Role of asymmetric dimethylarginine in acute lung injury induced by cerebral ischemia/reperfusion injury in rats. Nan Fang Yi Ke Da Xue Xue Bao 2011, 31, 1289–1294. [Google Scholar]
- Liu, Y.Z.; Zhou, Y.; Li, D.; Wang, L.; Hu, G.Y.; Peng, J.; Li, Y.J. Reduction of asymmetric dimethylarginine in the protective effects of rutaecarpine on gastric mucosal injury. Can. J. Physiol. Pharmacol. 2008, 86, 675–681. [Google Scholar]
- Chen, Q.Q.; Li, D.; Guo, R.; Luo, D.; Yang, J.; Hu, C.P.; Li, Y.J. Decrease in the synthesis and release of calcitonin gene-related peptide in dorsal root ganglia of spontaneously hypertensive rat: Role of nitric oxide synthase inhibitors. Eur J Pharmacol 2008, 596, 132–137. [Google Scholar]
- Lu, R.; Zhu, H.Q.; Peng, J.; Li, N.S.; Li, Y.J. Endothelium-dependent vasorelaxation and the expression of calcitonin gene-related peptide in aged rats. Neuropeptides 2002, 36, 407–412. [Google Scholar]
- Li, D.; Chen, B.M.; Peng, J.; Zhang, Y.S.; Li, X.H.; Yuan, Q.; Hu, C.P.; Deng, H.W.; Li, Y.J. Role of anandamide transporter in regulating calcitonin gene-related peptide production and blood pressure in hypertension. J Hypertens 2009, 27, 1224–1232. [Google Scholar]
- Brzozowski, T.; Konturek, P.C.; Sliwowski, Z.; Pajdo, R.; Drozdowicz, D.; Kwiecien, S.; Burnat, G.; Konturek, S.J.; Pawlik, W.W. Prostaglandin/cyclooxygenase pathway in ghrelin-induced gastroprotection against ischemia-reperfusion injury. J. Pharmacol. Exp. Ther. 2006, 319, 477–487. [Google Scholar]
- Wada, K.; Kamisaki, Y.; Kitano, M.; Kishimoto, Y.; Nakamoto, K.; Itoh, T. A new gastric ulcer model induced by ischemia-reperfusion in the rat: Role of leukocytes on ulceration in rat stomach. Life Sci. 1996, 59, 295–301. [Google Scholar]
- Konturek, S.J.; Brzozowski, T.; Pytko-Polonczyk, J.; Drozdowicz, D. Exogenous and endogenous cholecystokinin protects gastric mucosa against the damage caused by ethanol in rats Eur. J. Pharmacol. 1995, 273, 57–62. [Google Scholar]
- Konturek, P.C.; Brzozowski, T.; Sliwowski, Z.; Pajdo, R.; Stachura, J.; Hahn, E.G.; Konturek, S.J. Involvement of nitric oxide and prostaglandins in gastroprotection induced by bacterial lipopolysaccharide. Scand. J. Gastroenterol. 1998, 33, 691–700. [Google Scholar]
- Green, L.C.; Tannenbaum, S.R.; Goldman, P. Nitrate synthesis in the germ-free and conventional rat. Science 1981, 32, 56–58. [Google Scholar]
- Ignarro, L.J. Nitric oxide-mediated vasorelaxation. Tromb. Haemost. 1993, 32, 148–151. [Google Scholar]
- Brzozowski, T.; Konturek, P.C.; Sliwowski, Z.; Drozdowicz, D.; Burnat, G.; Pajdo, R.; Pawlik, M.; Bielanski, W.; Kato, I.; Kuwahara, A.; et al. Gastroprotective action of orexin-A against stress-induced gastric damage is mediated by endogenous prostaglandins sensory afferent neuropeptides and nitric oxide. Regul. Pept. 2008, 148, 6–20. [Google Scholar]
- Peng, J.; Lu, R.; Deng, H.W.; Li, Y.J. Involvement of alpha-calcitonin gene-related peptide in monophosphoryl lipid A-induced delayed preconditioning in rat hearts. Eur. J. Pharmacol. 2002, 436, 89–96. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental group | Plasma ADMA concentration (μmol/L) |
|---|---|
| Vehicle | 1.46 ± 0.22 |
| ADMA (0.1 mg/kg i.g.) | 1.83 ± 0.36 |
| ADMA (1 mg/kg i.g.) | 2.37 ± 0.46 * |
| ADMA (10 mg/kg i.g.) | 2.65 ± 0.34 * |
| ADMA (20 mg/kg i.g.) | 3.62 ± 0.43 *+ |
| ADMA (40 mg/kg i.g.) | 4.95 ± 0.58 *+ |
| Experimental group | Luminal NO (μmol/L) |
|---|---|
| After ischemia before reperfusion | |
| Vehicle | 15.2 ± 2.06 |
| ADMA (20 mg/kg i.g.) | 7.6 ± 0.08 * |
| After ischemia/reperfusion | |
| Vehicle | 14.6 ± 1.35 |
| ADMA (20 mg/kg i.g.) | 6.3 ± 0.04 * |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Magierowski, M.; Jasnos, K.; Sliwowski, Z.; Surmiak, M.; Krzysiek-Maczka, G.; Ptak-Belowska, A.; Kwiecien, S.; Brzozowski, T. Exogenous Asymmetric Dimethylarginine (ADMA) in Pathogenesis of Ischemia-Reperfusion-Induced Gastric Lesions: Interaction with Protective Nitric Oxide (NO) and Calcitonin Gene-Related Peptide (CGRP). Int. J. Mol. Sci. 2014, 15, 4946-4964. https://doi.org/10.3390/ijms15034946
Magierowski M, Jasnos K, Sliwowski Z, Surmiak M, Krzysiek-Maczka G, Ptak-Belowska A, Kwiecien S, Brzozowski T. Exogenous Asymmetric Dimethylarginine (ADMA) in Pathogenesis of Ischemia-Reperfusion-Induced Gastric Lesions: Interaction with Protective Nitric Oxide (NO) and Calcitonin Gene-Related Peptide (CGRP). International Journal of Molecular Sciences. 2014; 15(3):4946-4964. https://doi.org/10.3390/ijms15034946
Chicago/Turabian StyleMagierowski, Marcin, Katarzyna Jasnos, Zbigniew Sliwowski, Marcin Surmiak, Gracjana Krzysiek-Maczka, Agata Ptak-Belowska, Slawomir Kwiecien, and Tomasz Brzozowski. 2014. "Exogenous Asymmetric Dimethylarginine (ADMA) in Pathogenesis of Ischemia-Reperfusion-Induced Gastric Lesions: Interaction with Protective Nitric Oxide (NO) and Calcitonin Gene-Related Peptide (CGRP)" International Journal of Molecular Sciences 15, no. 3: 4946-4964. https://doi.org/10.3390/ijms15034946
APA StyleMagierowski, M., Jasnos, K., Sliwowski, Z., Surmiak, M., Krzysiek-Maczka, G., Ptak-Belowska, A., Kwiecien, S., & Brzozowski, T. (2014). Exogenous Asymmetric Dimethylarginine (ADMA) in Pathogenesis of Ischemia-Reperfusion-Induced Gastric Lesions: Interaction with Protective Nitric Oxide (NO) and Calcitonin Gene-Related Peptide (CGRP). International Journal of Molecular Sciences, 15(3), 4946-4964. https://doi.org/10.3390/ijms15034946