Modeling and Docking Studies on Novel Mutants (K71L and T204V) of the ATPase Domain of Human Heat Shock 70 kDa Protein 1

Abstract
:1. Introduction
2. Results and Discussion
2.1. In-Silico Mutagenesis
2.2. Physiochemical Characterization
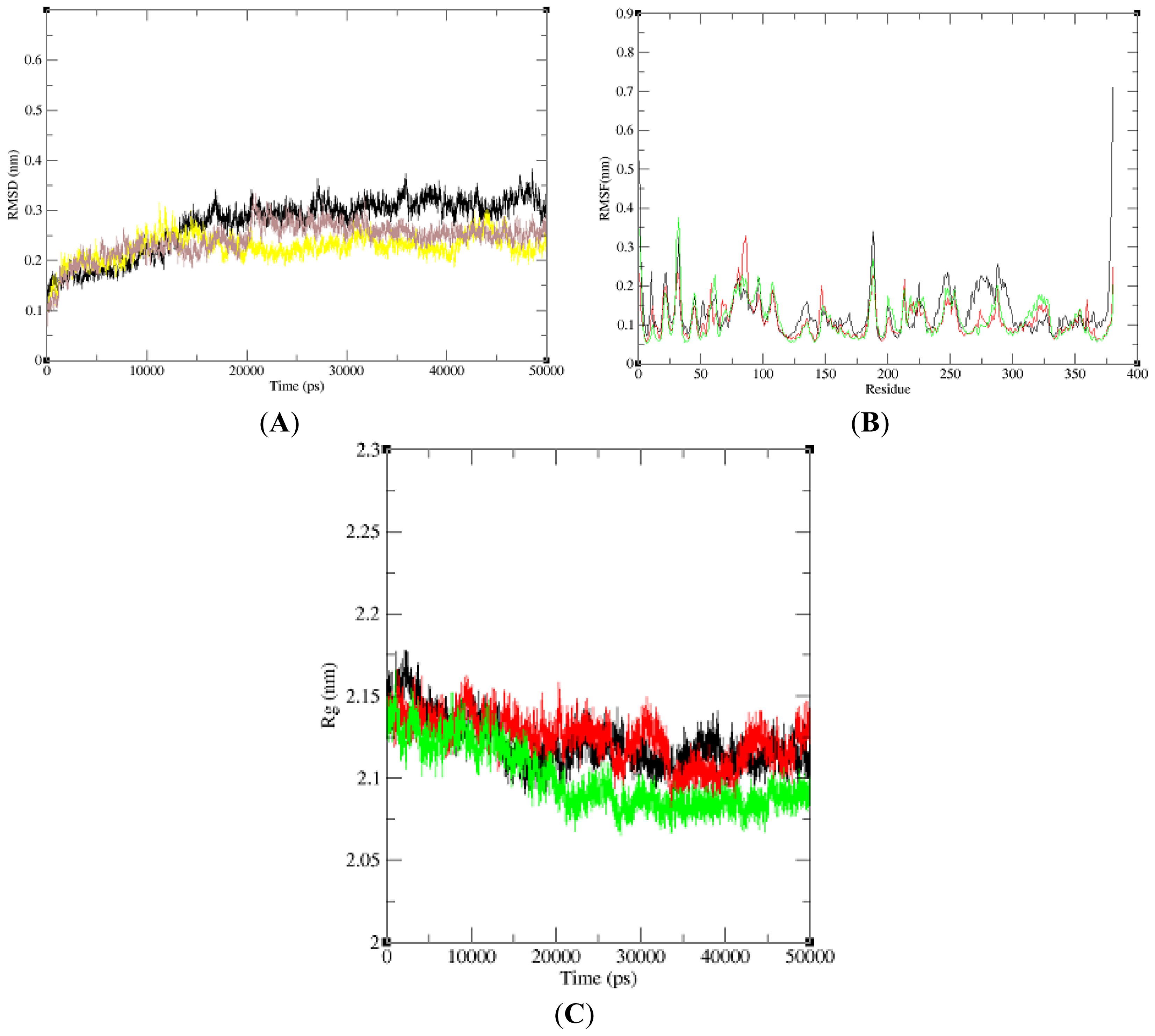
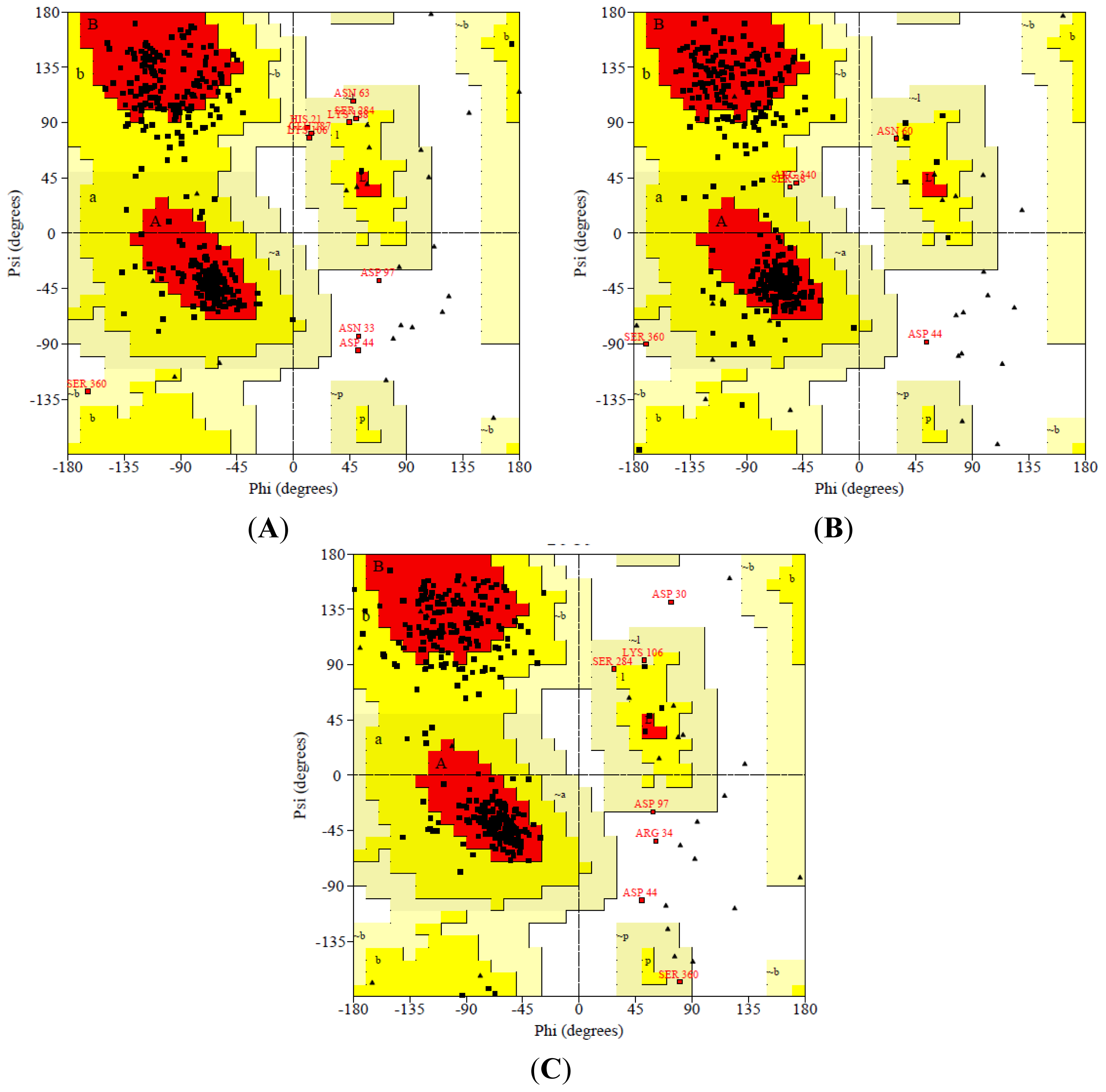
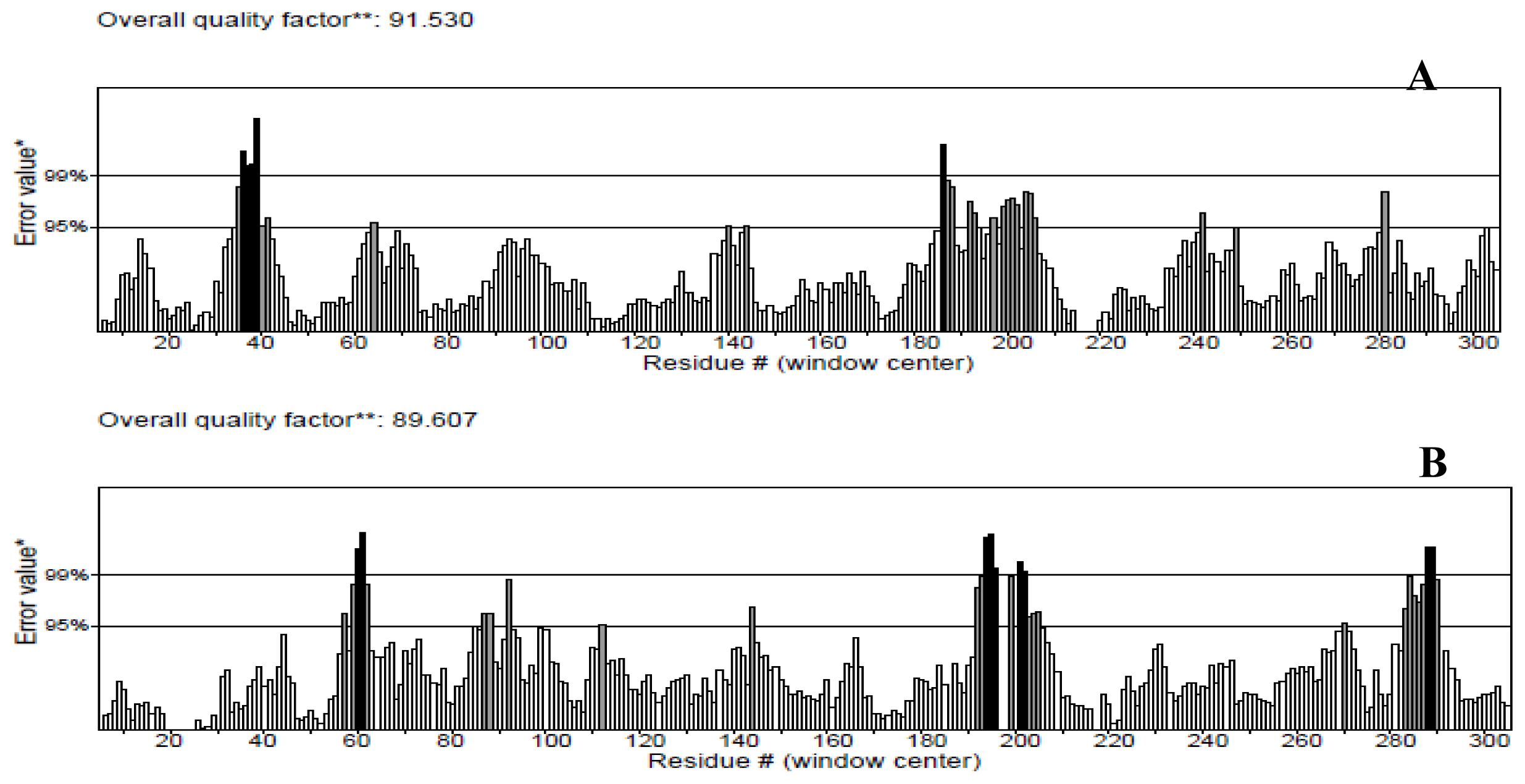
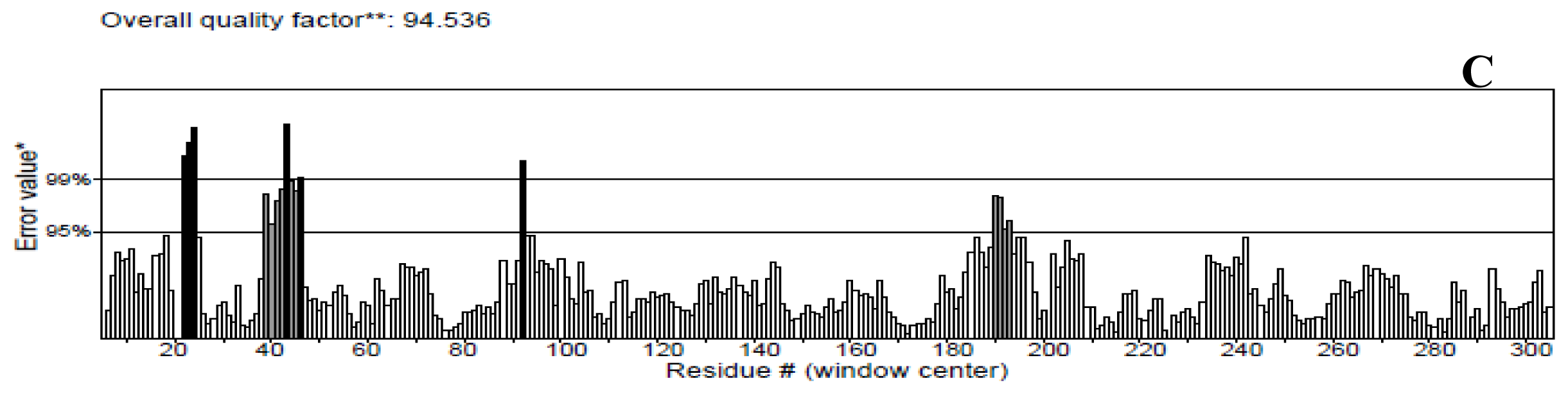
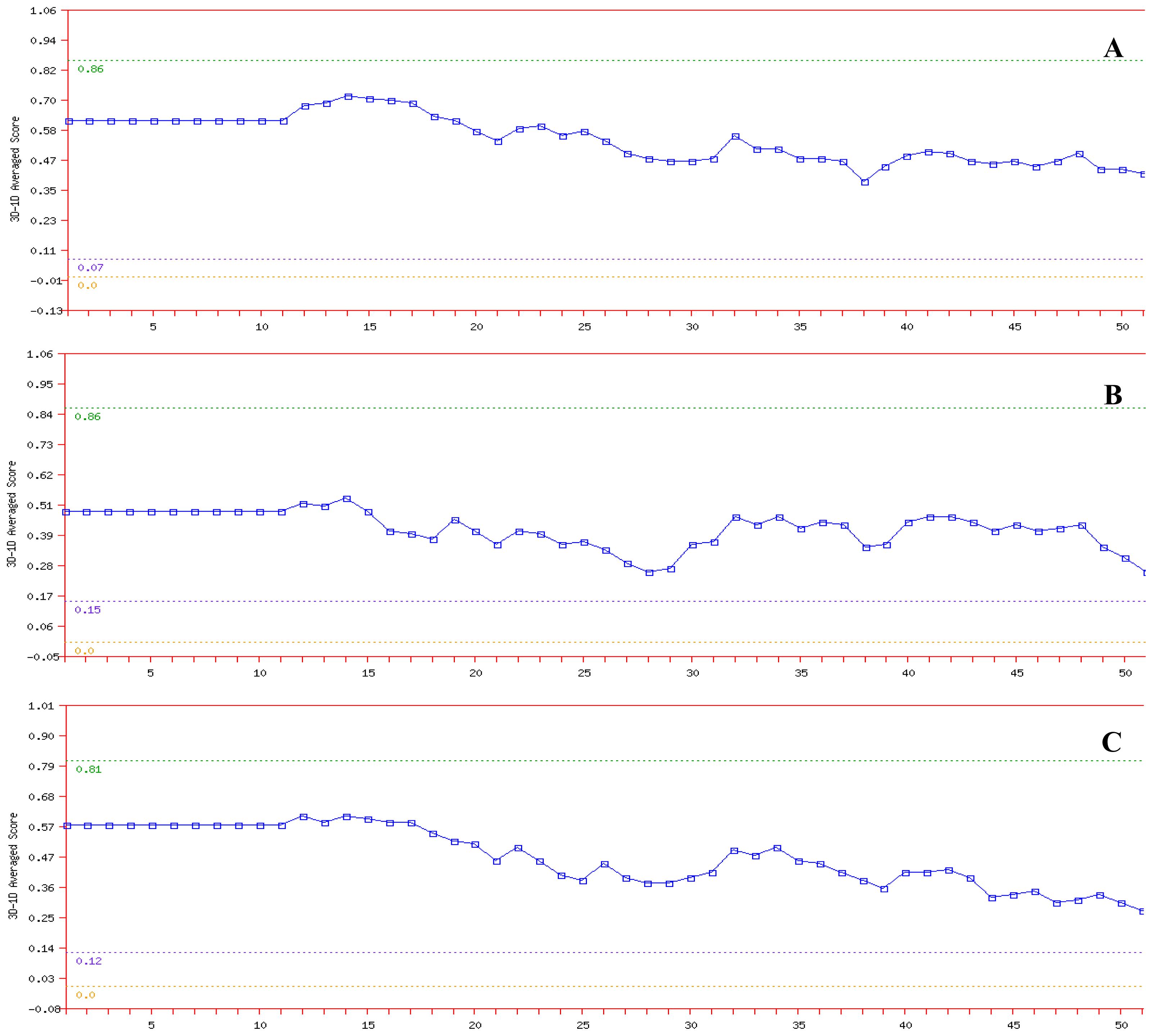
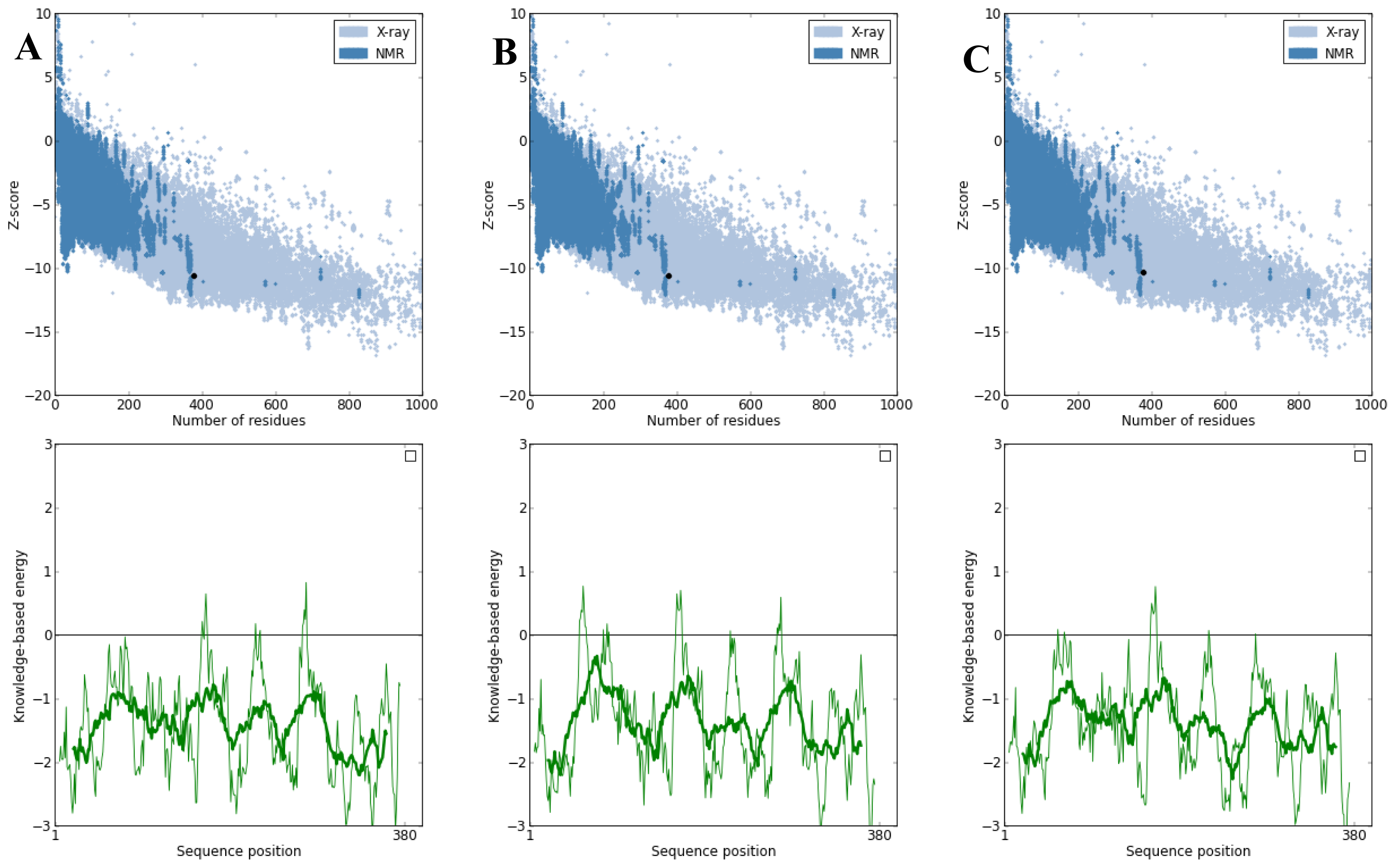
2.3. Model Simulation and Evaluation
2.4. Active Site Identification
2.5. Molecular Docking
2.6. Model Simulation and Evaluation of Protein-Ligand Complex
3. Experimental Section
3.1. Target Sequence
3.2. In-Silico Mutagenesis
3.3. Physiochemical Characterization
3.4. Model Simulation and Evaluation
3.5. Active Site Identification
3.6. Homology Modeling
3.7. Molecular Docking
3.8. Molecular Dynamics Simulation of Protein-Ligand Complex
4. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsAE designed the research, performed the experiments, analysed data and wrote the paper. MAN and SH contributed critical comments and suggestions. All authors read and approved the final manuscript.
References
- Hightower, L.E. Heat shock, stress proteins, chaperones, and proteotoxicity. Cell 1991, 66, 191–197. [Google Scholar]
- Gething, M.J.; Sambrook, J. Protein folding in the cell. Nature 1992, 355, 33–45. [Google Scholar]
- Netzer, W.J.; Hartl, F.U. Protein folding in the cytosol: Chaperon independent and -independent mechanisms. Trends Biochem. Sci 1998, 23, 68–73. [Google Scholar]
- Freeman, B.C.; Yamamoto, K.R. Disassembly of transcriptional regulatory complexes by molecular chaperones. Science 2002, 296, 2232–2235. [Google Scholar]
- Thorne, S.H.; Brooks, G.; Lee, Y.L.; Au, T.; Eng, L.F.; Reid, T. Effects of febrile temperature on adenoviral infection and replication: Implications for viral therapy of cancer. J. Virol 2005, 79, 581–591. [Google Scholar]
- Kao, H.T.; Capasso, O.; Heintz, N.; Nevins, J.R. Cell cycle control of the human HSP70 gene: Implications for the role of a cellular E1A-like function. Mol. Cell. Biol 1985, 5, 628–633. [Google Scholar]
- Wickner, S.; Skowyra, D.; Hoskins, J.; Mckenney, K. DnaJ, DnaK, and GrpE heat shock proteins are required in oriP1 DNA replication solely at the RepA monomerization step. Proc. Natl. Acad. Sci. USA 1992, 89, 10345–10349. [Google Scholar]
- Glotzer, J.B.; Saltik, M.; Chiocca, S.; Michou, A.I.; Moseley, P.; cotton, M. Activation of heat-shock response by an adenovirus is essential for virus replication. Nature 2000, 407, 207–211. [Google Scholar]
- Freeman, B.C.; Myers, M.P.; Schumacher, R.; Morimoto, R.I. Identification of a regulatory motif in Hsp70 that affects ATPase activity, substrate binding and interaction with HDJ-1. EMBO J 1995, 14, 2281–2292. [Google Scholar]
- Freeman, B.C.; Morimoto, R.I. The human cytosolic molecular chaperones Hsp90, Hsp70 (Hsc70), and HDJ-1 have distinct roles in recognition of a non-native protein and protein refolding. EMBO J 1996, 15, 2969–2979. [Google Scholar]
- Biro, J.C.; Benyo, B.; Sansom, C.; Szlavecz, A.; Fordos, G.; Micsik, T.; Benyo, Z. A common periodic table of codons and amino acids. Biochem. Biophys. Res. Commun 2003, 306, 408–415. [Google Scholar]
- O’Brien, M.C.; Flaherty, K.M.; McKay, D.B. Lysine 71 of the chaperone protein Hsc70 is essential for ATP hydrolysis. J. Biol. Chem 1996, 271, 15874–15878. [Google Scholar]
- Flaherty, K.M.; Wilbanks, S.M.; DeLuca-Flaherty, C.; McKay, D.B. Structural basis of the 70 kilodalton heat shock cognate protein ATP hydrolytic activity. J. Biol. Chem 1994, 269, 12899–12907. [Google Scholar]
- Wilbanks, S.M.; McKay, D.B. How potassium affects the activity of the molecular chaperone Hsc70. J. Biol. Chem 1995, 270, 2251–2257. [Google Scholar]
- McCarty, J.S.; Walker, G.C. DnaK as a thermometer: Threonine-199 is site of autophosphorylation and is critical for ATPase activity. Proc. Natl. Acad. Sci. USA 1991, 88, 9513–9517. [Google Scholar]
- Buchberger, A.; Schroder, H.; Buttner, M.; Valencia, A.; Bukau, B. A conserved loop in the ATPase domain of the DnaK chaperone is essential for stable binding of GrpE. Nat. Struct. Biol 1994, 1, 95–101. [Google Scholar]
- Wawrzynow, A.; Banecki, B.; Wall, D.; Liberek, K.; Georgopoulos, C. ATP hydrolysis is required for the DnaJ-dependent activation of DnaK chaperone for binding to both native and denatured protein substrates. J. Biol. Chem 1995, 270, 19307–19311. [Google Scholar]
- Campbell, K.S.; Mullane, K.P.; Aksoy, I.A.; Stubdal, H.; Zalvide, J.; Pipas, J.M.; Silver, P.A.; Roberts, T.M.; Schaffhausen, B.S.; DeCaprio, J.A. DnaJ/hsp40 chaperone domain of SV40 large T antigen promotes efficient viral DNA replication. Genes Dev 1997, 11, 1098–1110. [Google Scholar]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005. [Google Scholar]
- Geourjon, C.; Deleage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci 1995, 11, 681–684. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst 1993, 26, 283–291. [Google Scholar]
- Reddy, Ch.S.; Vijayasarathy, K.; Srinivas, E.; Sastry, G.M.; Sastry, G.N. Homology modeling of membrane proteins: A critical assessment. Comput. Biol. Chem 2006, 30, 120–126. [Google Scholar]
- Gundampati, R.K.; Chikati, R.; Kumari, M.; Sharma, A.; Pratyush, D.D.; Jagannadham, M.V.; Kumar, C.S.; Das, M.D. Protein-protein docking on molecular models of Aspergillus niger RNase and human actin: Novel target for anticancer therapeutics. J. Mol. Model 2012, 18, 653–662. [Google Scholar]
- Wallner, B.; Elofsson, A. Can correct protein models be identified? Protein Sci 2003, 12, 1073–1086. [Google Scholar]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of non-bonded atomic interactions. Protein Sci 1993, 2, 1511–1519. [Google Scholar]
- Eisenberg, D.; Luthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol 1997, 277, 396–404. [Google Scholar]
- Wiederstein, M.; Sippl, M. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 2007, 35, W407–W410. [Google Scholar]
- Steiner, T.; Koellner, G. Hydrogen bonds with p–acceptors in proteins: Frequencies and role in stabilizing local 3–D structures. J. Mol. Biol 2001, 305, 535–557. [Google Scholar]
- Priya, George; Doss, C.; Nagasundaram, N. Investigating the structural impacts of I64T and P311S mutations in APE1-DNA complex: A Molecular Dynamics Approach. PLoS One 2012, 7, 1–11. [Google Scholar]
- Weiss, M.S.; Brandl, M.; Sühnel, J.; Pal, D.; Hilgenfeld, R. More hydrogen bonds for the (structural) biologist. Trends Biochem. Sci 2001, 26, 521–523. [Google Scholar]
- Delano, W.L. The PyMOL Molecular Graphics System, 2002. 10 September 2013. Available online: http://www.pymol.org.
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem 2005, 26, 1701–1718. [Google Scholar]
- Laurie, A.T.; Jackson, R.M. Q-SiteFinder: An energy-based method for the prediction of protein-ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [Google Scholar]
- Burgoyne, N.J.; Jackson, R.M. Predicting protein interaction sites: Binding hot-spots in protein-protein and protein-ligand interfaces. Bioinformatics 2006, 22, 1335–1342. [Google Scholar]
- Kuntal, B.K.; Aparoy, P.; Reddanna, P. EasyModeller: A graphical interface to MODELLER. BMC Res. Notes 2010, 3, 226. [Google Scholar]
- Fiser, A.; Sali, A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol 2003, 374, 461–491. [Google Scholar]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model 1999, 17, 57–61. [Google Scholar]
- Schuttelkopf, A.W.; van Aalten, D.M. PRODRG—A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr 2004, 60, 1355–1363. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Length | M.wt (Daltons) | pI | −R | +R | Extinction coefficient (M−1·cm−1) | Instability index | Aliphatic index | GRAVY |
|---|---|---|---|---|---|---|---|---|---|
| NBD | 380 | 41,827.4 | 6.69 | 50 | 49 | 20,525 | 35.09 | 88.32 | −0.274 |
| K71L | 380 | 41,812.4 | 6.38 | 50 | 48 | 20,525 | 34.74 | 89.34 | −0.253 |
| T204V | 380 | 41,825.5 | 6.69 | 50 | 49 | 20,525 | 34.99 | 89.08 | −0.261 |
| Secondary structure | Alpha helix(Hh) | Extended strand (Ee) | Beta turn (Tt) | Random coil (Cc) |
|---|---|---|---|---|
| NBD | 42.89 | 19.74 | 7.63 | 29.74 |
| K71L | 44.21 | 18.68 | 8.16 | 28.95 |
| T204V | 44.47 | 18.95 | 6.84 | 29.74 |
| Structure | Ramachandran plot statistics (%) | Goodness factor | ProQ | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Most favoured | Additionally allowed | Generously allowed | Disallowed | Dihedral angles | Covalent forces | Overall average | LG Score | Max–sub | |
| NBD | 81.7 | 15.4 | 2.1 | 0.9 | −0.61 * | −0.95 * | −0.66 * | 5.707 | 0.451 |
| K71L | 79.9 | 18.6 | 0.9 | 0.6 | −0.59 * | −1.02 ** | −0.67 * | 5.497 | 0.425 |
| T204V | 78.7 | 19.2 | 1.2 | 0.9 | −0.61 * | −0.91 * | −0.64 * | 5.862 | 0.424 |
| Protein | Site volume (cubic Å) | Protein volume (cubic Å) | Residues that forming pocket |
|---|---|---|---|
| NBD | 434 | 34357 | ASP10,LEU11,GLY12,THR13,THR14,TYR15,PHE68,ASP69,LYS71,ARG72,TRP90,THR145,VAL146,PRO147,ALA148,GLU175,PRO176,ILE197,PHE198,ASP199,GLY201,GLY202,GLY203,THR204,ASP206,VAL207,SER208,THR222,VAL337,VAL369 |
| K71L | 496 | 34418 | ASP10,LEU11,GLY12,THR13,PRO14,TYR15,CYS17,ARG72,VAL146,PRO147,ALA148,TYR149,GLU175,PRO176,ALA179,ILE197,PHE198,ASP199,LEU200,GLY201,GLY202,GLY203,THR204,ASP206,VAL207,SER208,THR222,ALA223,GLY224,LYS271,ARG272,VAL337,GLY338,GLY339,GLY340,ALA368,VAL369,ALA370 |
| T204V | 532 | 34224 | ASP10,LEU11,GLY12,THR13,THR14,TYR15,SER16,CYS17,LYS71,ARG72,VAL82,THR145,PRO147,ALA148,TYR149,PHE150,GLU175,ASP199,LEU200,GLY201,GLY202,GLY203,VAL204,PHE205,ASP206,ARG272,VAL337,GLY338,GLY339,PRO365,ASP366,GLU367,ALA368,VAL369,ALA370 |
| Protein | NBD | K71L | T204V |
|---|---|---|---|
| Binding energy (kcal/mol) | −8.05 | −6.76 | −9.09 |
| kI (μM) | 1.26 | 11.04 | 0.22 |
| Intermolecular Energy (kcal/mol) | −11.93 | −10.64 | −12.97 |
| Internal energy (kcal/mol) | −2.45 | −2.55 | −1.49 |
| Torsion energy (kcal/mol) | 3.88 | 3.88 | 3.88 |
| Unbounded Extended energy (kcal/mol) | −2.45 | −2.55 | −1.49 |
| Cluster RMS | 0.00 | 0.00 | 0.00 |
| Reference RMS | 86.09 | 72.93 | 76.96 |
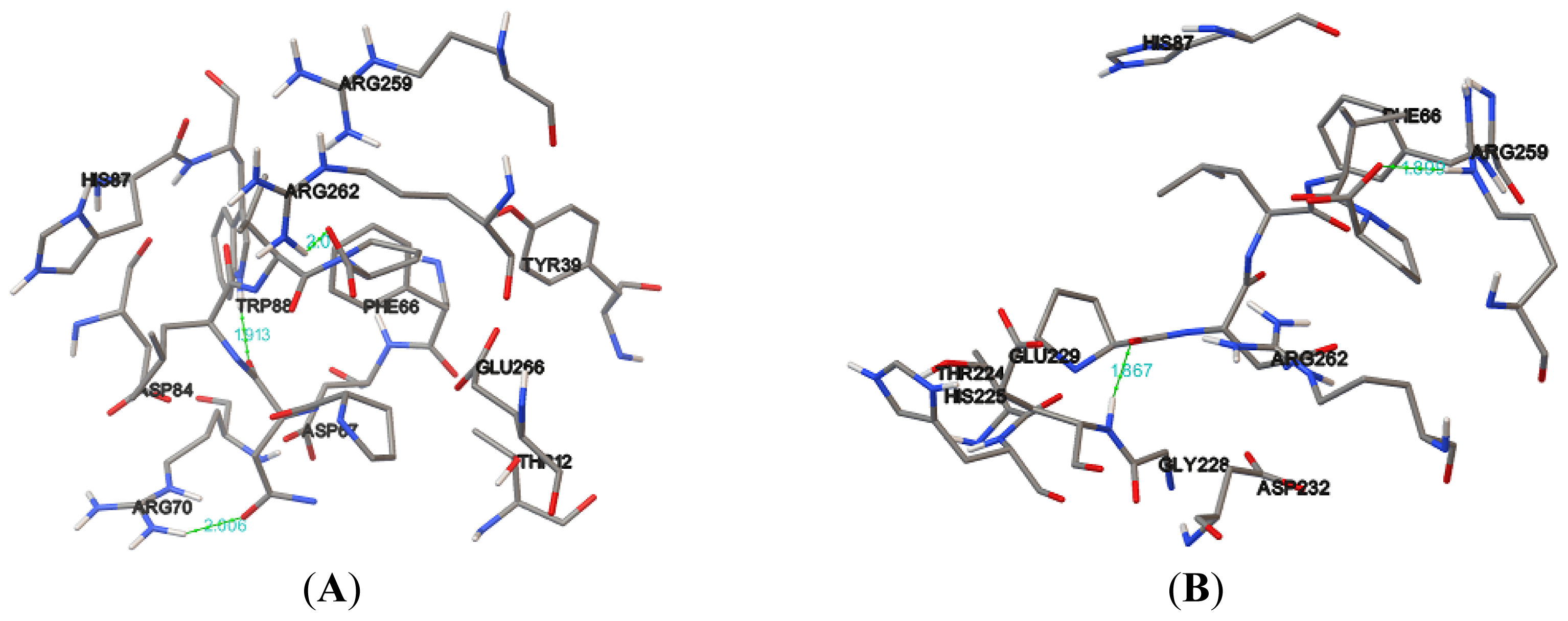
| Protein | Donor atom | Acceptor atom | Distance (Å) |
|---|---|---|---|
| NBD | ARG72:HH21 | ASN2:OD1 | 1.913 |
| TRP90:HE1 | ASN2:O | 2.006 | |
| ARG264:HH11 | PRO5:O,OXT | 2.000 | |
| K71L | GLU231:HN | PRO1:O | 1.867 |
| ARG261:HE | PRO5:O | 1.899 | |
| T204V | THR14:HN | PRO5:O | 2.065 |
| ARG72:HE | PRO:O | 2.199 | |
| ARG72:HH21 | ASN2:O | 1.594 | |
| THR13:HN | VAL4:O | 1.922 | |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Elengoe, A.; Naser, M.A.; Hamdan, S. Modeling and Docking Studies on Novel Mutants (K71L and T204V) of the ATPase Domain of Human Heat Shock 70 kDa Protein 1. Int. J. Mol. Sci. 2014, 15, 6797-6814. https://doi.org/10.3390/ijms15046797
Elengoe A, Naser MA, Hamdan S. Modeling and Docking Studies on Novel Mutants (K71L and T204V) of the ATPase Domain of Human Heat Shock 70 kDa Protein 1. International Journal of Molecular Sciences. 2014; 15(4):6797-6814. https://doi.org/10.3390/ijms15046797
Chicago/Turabian StyleElengoe, Asita, Mohammed Abu Naser, and Salehhuddin Hamdan. 2014. "Modeling and Docking Studies on Novel Mutants (K71L and T204V) of the ATPase Domain of Human Heat Shock 70 kDa Protein 1" International Journal of Molecular Sciences 15, no. 4: 6797-6814. https://doi.org/10.3390/ijms15046797
APA StyleElengoe, A., Naser, M. A., & Hamdan, S. (2014). Modeling and Docking Studies on Novel Mutants (K71L and T204V) of the ATPase Domain of Human Heat Shock 70 kDa Protein 1. International Journal of Molecular Sciences, 15(4), 6797-6814. https://doi.org/10.3390/ijms15046797