Association of Dioxin and Other Persistent Organic Pollutants (POPs) with Diabetes: Epidemiological Evidence and New Mechanisms of Beta Cell Dysfunction

{kind=link}
Abstract
:1. Introduction
2. Epidemiological Evidence
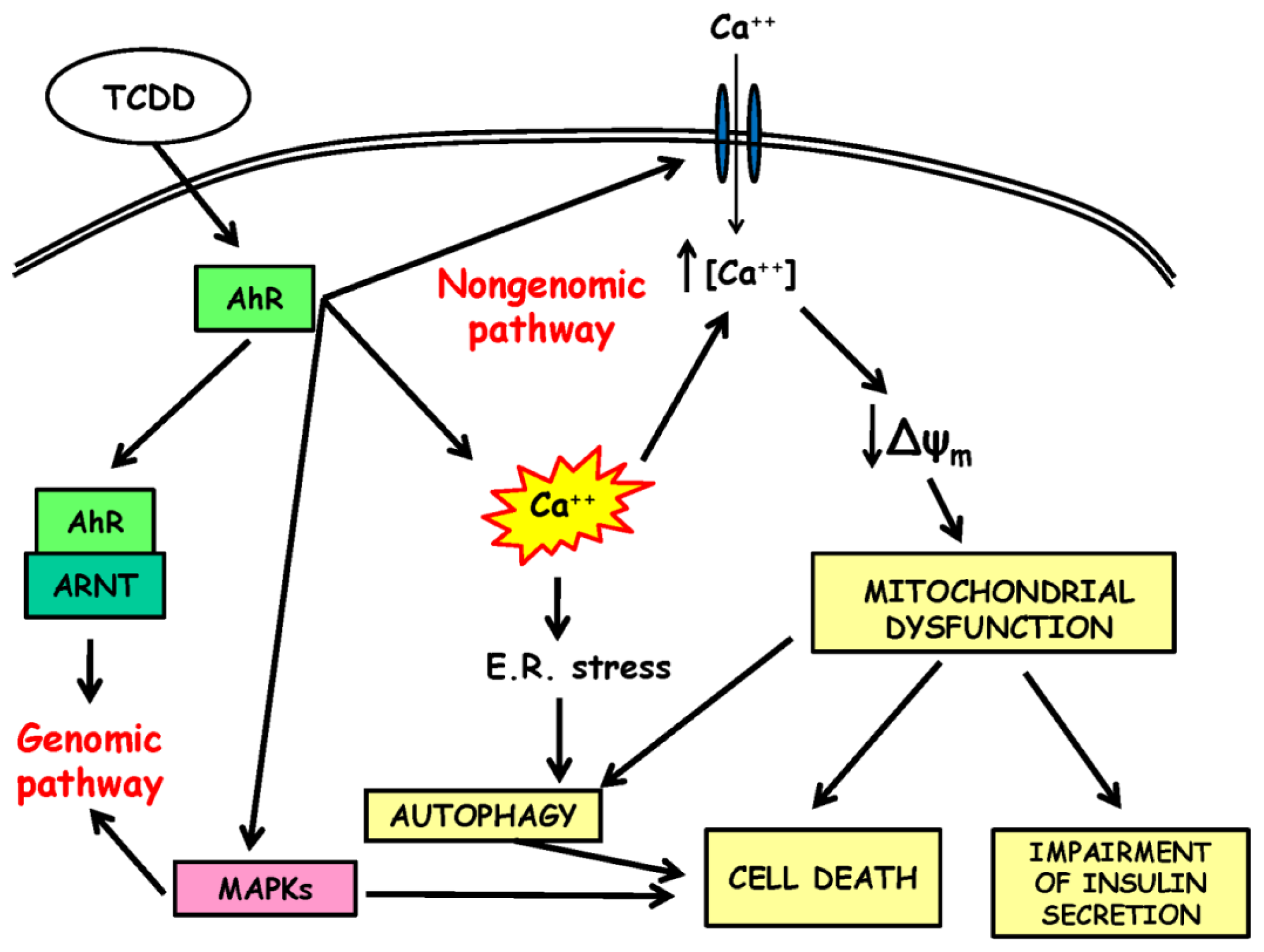
3. Mechanisms of Dioxin-Induced Beta Cell Dysfunction
3.1. In Vivo Effects of Dioxin Administration
3.2. The Non-Genomic Pathway of Dioxin Toxicity
3.3. Dioxin-Induced Mitochondrial Dysfunction
3.4. POP Exposure and Obesity
3.5. Dioxin-Induced Alteration of Signal Transduction Pathways
3.6. Dioxin-Induced Activation of Autophagy
4. Conclusions
Conflicts of Interest
References
- Wild, S.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care 2004, 27, 1047–1053. [Google Scholar]
- Kolb, H.; Mandrup-Poulsen, T. The global diabetes epidemics as a consequence of lifestyle-induced low-grade inflammation. Diabetologia 2010, 53, 10–20. [Google Scholar]
- Shaw, J.E.; Sicree, R.A.; Zimmet, P.Z. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract 2010, 87, 4–14. [Google Scholar]
- Longnecker, M.P.; Daniels, J.L. Environmental contaminants as etiologic factors for diabetes. Environ. Health Perspect 2001, 109, 871–876. [Google Scholar]
- Neel, B.A.; Sargis, R.M. The paradox of progress: Environmental disruption of metabolism and the diabetes epidemic. Diabetes 2011, 60, 1838–1848. [Google Scholar]
- Remillard, R.B.; Bunce, N.J. Linking dioxins to diabetes: Epidemiology and biologic plausibility. Environ. Health Perspect 2002, 110, 853–858. [Google Scholar]
- Environmental Protection Agency, Special Report on Environmental Endocrine Disruption: An Effects Assessment and Analysis; U.S Environmental Protection Agency: Washington, DC, USA, 1997.
- Müllerová, D.; Kopecký, J. White adipose tissue: Storage and effector site for environmental pollutants. Physiol. Res 2007, 56, 375–381. [Google Scholar]
- Schug, T.T.; Janesick, A.; Blumberg, B.; Heindel, J.J. Endocrine disrupting chemicals and disease susceptibility. J. Steroid Biochem. Mol. Biol 2011, 127, 204–215. [Google Scholar]
- Alonso-Magdalena, P.; Quesada, I.; Nadal, A. Endocrine disruptors in the etiology of type 2 diabetes mellitus. Nat. Rev. Endocrinol 2011, 7, 346–353. [Google Scholar]
- Schafer, K.S.; Kegley, S.E. Persistent toxic chemicals in the US food supply. J. Epidemiol. Commun. Health 2002, 56, 813–817. [Google Scholar]
- Arisawa, K.; Takeda, H.; Mikasa, H. Background exposure to PCDDs/PCDFs/PCBs and its potential health effects: A review of epidemiologic studies. J. Med. Investig 2005, 52, 10–21. [Google Scholar]
- Lee, D.-H.; Jacobs, D.R.; Porta, M. Editorial: Could low-level background exposure to persistent organic pollutants contribute to the social burden of type 2 diabetes? J. Epidemiol. Commun. Health 2008, 60, 1006–1008. [Google Scholar]
- Taylor, K.W.; Novak, R.F.; Anderson, H.A.; Birnbaum, L.S.; Blystone, C.; Devito, M.; Jacobs, D.; Köhrle, J.; Lee, D.H.; Rylander, L.; et al. Evaluation of the association between persistent organic pollutants (POPs) and diabetes in epidemiological studies: A national toxicology program workshop review. Environ. Health Perspect 2013, 121, 774–783. [Google Scholar]
- Magliano, D.J.; Loh, V.H.; Harding, J.L.; Botton, J.; Shaw, J.E. Persistent organic pollutants and diabetes: A review of the epidemiological evidence. Diabetes Metab 2014, 40, 1–14. [Google Scholar]
- Booker, S.M. Dioxin in Vietnam: Fighting a legacy of war. Environ. Health Perspect 2001, 109, A116–A117. [Google Scholar]
- Longnecker, M.P.; Michalek, J.E. Serum dioxin level in relation to diabetes mellitus a mong Air Force veterans with background levels of exposure. Epidemiology 2000, 11, 44–48. [Google Scholar]
- Pavuk, M.; Patterson, D.G., Jr.; Turner, W.E.; Needham, L.L.; Ketchum, N.S. Polychlorinated dibenzo-p-dioxins (PCDDs), polychlorinated dibenzofurans (PCDFs), and dioxin-like polychlorinated biphenyls (PCBs) in the serum of US Air Force veterans in 2002. Chemosphere 2007, 68, 62–68. [Google Scholar]
- Michalek, J.E.; Tripathi, R.C. Pharmacokinetics of TCDD in veterans of Operation Ranch Hand: 15-year follow-up. J. Toxicol. Environ. Health A 1999, 57, 369–378. [Google Scholar]
- Henriksen, G.L.; Ketchum, N.S.; Michalek, J.E.; Swaby, J.A. Serum dioxin and diabetes mellitus in veterans of Operation Ranch Hand. Epidemiology 1997, 8, 252–258. [Google Scholar]
- Michalek, J.E.; Akhtar, F.Z.; Kiel, J.L. Serum dioxin, insulin, fasting glucose, and sex hormone-binding globulin in veterans of Operation Ranch Hand. J. Clin. Endocrinol. Metab 1999, 84, 1540–1543. [Google Scholar]
- Michalek, J.E.; Ketchum, N.S.; Tripathi, R.C. Diabetes mellitus and 2,3,7,8-tetrachlorodibenzo-p-dioxin elimination in veterans of Operation Ranch Hand. J. Toxicol. Environ. Health A 2003, 66, 211–221. [Google Scholar]
- Kang, H.K.; Dalager, N.A.; Needham, L.L.; Patterson, D.G., Jr.; Lees, P.S.; Yates, K.; Matanoski, G.M. Health status of Army Chemical Corps Vietnam veterans who sprayed defoliant in Vietnam. Am. J. Ind. Med 2006, 49, 875–884. [Google Scholar]
- Michalek, J.E.; Pavuk, M. Diabetes and cancer in veterans of Operation Ranch Hand after adjustment for calendar period, days of spraying, and time spent in Southeast Asia. J. Occup. Environ. Med 2008, 50, 330–340. [Google Scholar]
- Bertazzi, P.A.; Bernucci, I.; Brambilla, G.; Consonni, D.; Pesatori, A.C. The Seveso studies on early and long-term effects of dioxin exposure: A review. Environ. Health Perspect 1998, 106, 625–633. [Google Scholar]
- Bertazzi, P.A.; di Domenico, A. Health consequences of the Seveso, Italy, accident. In Dioxin and Health, 2nd ed; Schecter, A., Gasiewicz, T.A., Eds.; Wiley: Hoboken, NJ, USA, 2003; pp. 827–853. [Google Scholar]
- Pesatori, A.C. Dioxin contamination in Seveso: The social tragedy and the scientific challenge. Med. Lav 1995, 86, 111–124. [Google Scholar]
- Consonni, D.; Pesatori, A.C.; Zocchetti, C.; Sindaco, R.; D’Oro, L.C.; Rubagotti, M.; Bertazzi, P.A. Mortality in a population exposed to dioxin after the Seveso, Italy, accident in 1976: 25 years of follow-up. Am. J. Epidemiol 2008, 167, 847–858. [Google Scholar]
- Warner, M.; Mocarelli, P.; Brambilla, P.; Wesselink, A.; Samuels, S.; Signorini, S.; Eskenazi, B. Diabetes, metabolic syndrome, and obesity in relation to serum dioxin concentrations: The Seveso women’s health study. Environ. Health Perspect 2013, 121, 906–911. [Google Scholar]
- Naville, D.; Pinteur, C.; Vega, N.; Menade, Y.; Vigier, M.; le Bourdais, A.; Labaronne, E.; Debard, C.; Luquain-Costaz, C.; Bégeot, M.; et al. Low-dose food contaminants trigger sex-specific, hepatic metabolic changes in the progeny of obese mice. FASEB J 2013, 27, 3860–3870. [Google Scholar]
- Vena, J.; Boffetta, P.; Becher, H.; Benn, T.; Bueno-de-Mesquita, H.B.; Coggon, D.; Colin, D.; Flesch-Janys, D.; Green, L.; Kauppinen, T.; et al. Exposure to dioxin and nonneoplastic mortality in the expanded IARC international cohort study of phenoxy herbicide and chlorophenol production workers and sprayers. Environ. Health Perspect 1998, 106, 645–653. [Google Scholar]
- Kim, J.S.; Lim, H.S.; Cho, S.I.; Cheong, H.K.; Lim, M.K. Impact of Agent Orange exposure among Korean Vietnam veterans. Ind. Health 2003, 41, 149–157. [Google Scholar]
- Chen, H.L.; Su, H.J.; Guo, Y.L.; Liao, P.C.; Hung, C.F.; Lee, C.C. Biochemistry examinations and health disorder evaluation of Taiwanese living near incinerators and with low serum PCDD/Fs levels. Sci. Total Environ 2006, 366, 538–548. [Google Scholar]
- Kouznetsova, M.; Huang, X.; Ma, J.; Lessner, L.; Carpenter, D.O. Increased rate of hospitalization for diabetes and residential proximity of hazardous waste sites. Environ. Health Perspect 2007, 115, 75–79. [Google Scholar]
- Wang, S.L.; Tsai, P.C.; Yang, C.Y.; Leon Guo, Y. Increased risk of diabetes and polychlorinated biphenyls and dioxins: A 24-year follow-up study of the Yucheng cohort. Diabetes Care 2008, 31, 1574–1579. [Google Scholar]
- Steenland, K.; Piacitelli, L.; Deddens, J.; Fingerhut, M.; Chang, L.I. Cancer, heart disease, and diabetes in workers exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Natl. Cancer Inst 1999, 91, 779–786. [Google Scholar]
- Calvert, G.M.; Sweeney, M.H.; Deddens, J.; Wall, D.K. Evaluation of diabetes mellitus, serum glucose, and thyroid function among United States workers exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Occup. Environ. Med 1999, 56, 270–276. [Google Scholar]
- Steenland, K.; Calvert, G.; Ketchum, N.; Michalek, J. Dioxin and diabetes mellitus: An analysis of the combined NIOSH and Ranch Hand data. Occup. Environ. Med 2001, 58, 641–648. [Google Scholar]
- Karouna-Renier, N.K.; Rao, K.R.; Lanza, J.J.; Davis, D.A.; Wilson, P.A. Serum profiles of PCDDs and PCDFs, in individuals near the Escambia Wood Treating Company Superfund site in Pensacola, FL. Chemosphere 2007, 69, 1312–1319. [Google Scholar]
- Collins, J.J.; Bodner, K.; Aylward, L.L.; Wilken, M.; Bodnar, C.M. Mortality rates among trichlorophenol workers with exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Am. J. Epidemiol 2009, 170, 501–506. [Google Scholar]
- Collins, J.J.; Bodner, K.; Aylward, L.L.; Wilken, M.; Swaen, G.; Budinsky, R.; Rowlands, C.; Bodnar, C.M. Mortality rates among workers exposed to dioxins in the manufacture of pentachlorophenol. J. Occup. Environ. Med 2009, 51, 1212–1219. [Google Scholar]
- Kerger, B.D.; Scott, P.K.; Pavuk, M.; Gough, M.; Paustenbach, D.J. Re-analysis of Ranch Hand study supports reverse causation hypothesis between dioxin and diabetes. Crit. Rev. Toxicol 2012, 42, 669–687. [Google Scholar]
- Lee, D.H.; Porta, M.; Jacobs, D.R., Jr.; Vandenberg, L.N. Chlorinated persistent organic pollutants, obesity, and type 2 diabetes. Endocr. Rev 2014, in press. [Google Scholar]
- Fierens, S.; Mairesse, H.; Heilier, J.F.; de Burbure, C.; Focant, J.F.; Eppe, G.; de Pauw, E.; Bernard, A. Dioxin/polychlorinated biphenyl body burden, diabetes and endometriosis: findings in a population-based study in Belgium. Biomarkers 2003, 8, 529–534. [Google Scholar]
- Lee, D.H.; Lee, I.K.; Song, K.; Steffes, M.; Toscano, W.; Baker, B.A.; Jacobs, D.R., Jr. A strong dose-response relation between serum concentrations of persistent organic pollutants and diabetes: Results from the National Health and Examination Survey 1999–2002. Diabetes Care 2006, 29, 1638–1644. [Google Scholar]
- Lee, D.H.; Lee, I.K.; Steffes, M.; Jacobs, D.R., Jr. Extended analyses of the association between serum concentrations of persistent organic pollutants and diabetes. Diabetes Care 2007, 30, 1596–1598. [Google Scholar]
- Lee, D.H.; Lee, I.K.; Jin, S.H.; Steffes, M.; Jacobs, D.R., Jr. Association between serum concentrations of persistent organic pollutants and insulin resistance among nondiabetic adults: Results from the National Health and Nutrition Examination Survey 1999–2002. Diabetes Care 2007, 30, 622–628. [Google Scholar]
- Lee, D.H.; Lee, I.K.; Porta, M.; Steffes, M.; Jacobs, D.R., Jr. Relationship between serum concentrations of persistent organic pollutants and the prevalence of metabolic syndrome among non-diabetic adults: Results from the National Health and Nutrition Examination Survey 1999–2002. Diabetologia 2007, 50, 1841–1851. [Google Scholar]
- Lee, D.H.; Lind, L.; Jacobs, D.R., Jr.; Salihovic, S.; van Bavel, B.; Lind, P.M. Associations of persistent organic pollutants with abdominal obesity in the elderly: The Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) study. Environ. Int 2012, 40, 170–178. [Google Scholar]
- Everett, C.J.; Frithsen, I.L.; Diaz, V.A.; Koopman, R.J.; Simpson, W.M., Jr.; Mainous, A.G., 3rd. Association of a polychlorinated dibenzo-p-dioxin, a polychlorinated biphenyl, and DDT with diabetes in the 1999–2002 National Health and Nutrition Examination Survey. Environ. Res 2007, 103, 413–418. [Google Scholar]
- Lee, D.H.; Jacobs, D.R., Jr.; Gross, M.; Kiefe, C.I.; Roseman, J.; Lewis, C.E.; Steffes, M. Gamma-glutamyltransferase is a predictor of incident diabetes and hypertension: The Coronary Artery Risk Development in Young Adults (CARDIA) Study. Clin. Chem 2003, 49, 1358–1366. [Google Scholar]
- Lee, D.H.; Jacobs, D.R., Jr. Association between serum concentrations of persistent organic pollutants and gamma glutamyltransferase: Results from the National Health and Examination Survey 1999–2002. Clin. Chem 2006, 52, 1825–1827. [Google Scholar]
- Rignell-Hydbom, A.; Rylander, L.; Hagmar, L. Exposure to persistent organochlorine pollutants and type 2 diabetes mellitus. Hum. Exp. Toxicol 2007, 26, 447–452. [Google Scholar]
- Uemura, H.; Arisawa, K.; Hiyoshi, M.; Satoh, H.; Sumiyoshi, Y.; Morinaga, K.; Kodama, K.; Suzuki, T.; Nagai, M.; Suzuki, T. Associations of environmental exposure to dioxins with prevalent diabetes among general inhabitants in Japan. Environ. Res 2008, 108, 63–68. [Google Scholar]
- Rignell-Hydbom, A.; Lidfeldt, J.; Kiviranta, H.; Rantakokko, P.; Samsioe, G.; Agardh, C.D.; Rylander, L. Exposure to p,p′-DDE: A risk factor for type 2 diabetes. PLoS One 2009, 4, e7503. [Google Scholar]
- Lee, D.H.; Steffes, M.W.; Sjödin, A.; Jones, R.S.; Needham, L.L.; Jacobs, D.R., Jr. Low dose of some persistent organic pollutants predicts type 2 diabetes: A nested case-control study. Environ. Health Perspect 2010, 118, 1235–1242. [Google Scholar]
- Tanaka, T.; Morita, A.; Kato, M.; Hirai, T.; Mizoue, T.; Terauchi, Y.; Watanabe, S.; Noda, M. SCOP Study Group. Congener-specific polychlorinated biphenyls and the prevalence of diabetes in the Saku Control Obesity Program (SCOP). Endocr. J 2011, 58, 589–596. [Google Scholar]
- Lee, D.H.; Lind, P.M.; Jacobs, D.R., Jr.; Salihovic, S.; van Bavel, B.; Lind, L. Polychlorinated biphenyls and organochlorine pesticides in plasma predict development of type 2 diabetes in the elderly: The prospective investigation of the vasculature in Uppsala Seniors (PIVUS) study. Diabetes Care 2011, 34, 1778–1784. [Google Scholar]
- Everett, C.J.; Thompson, O.M. Associations of dioxins, furans and dioxin-like PCBs with diabetes and pre-diabetes: Is the toxic equivalency approach useful? Environ. Res 2012, 118, 107–111. [Google Scholar]
- Nakamoto, M.; Arisawa, K.; Uemura, H.; Katsuura, S.; Takami, H.; Sawachika, F.; Yamaguchi, M.; Juta, T.; Sakai, T.; Toda, E.; et al. Association between blood levels of PCDDs/PCDFs/dioxin-like PCBs and history of allergic and other diseases in the Japanese population. Int. Arch. Occup. Environ. Health 2013, 86, 849–859. [Google Scholar]
- Bonefeld-Jorgensen, E. Biomonitoring in Greenland: Human biomarkers of exposure and effects—A short review. Rural Remote Health 2010, 10, 1362. [Google Scholar]
- Jørgensen, M.E.; Borch-Johnsen, K.; Bjerregaard, P. A cross-sectional study of the association between persistent organic pollutants and glucose intolerance among Greenland Inuit. Diabetologia 2008, 51, 1416–1422. [Google Scholar] [Green Version]
- Sharp, D. Environmental toxins, a potential risk factor for diabetes among Canadian Aboriginals. Int. J. Circumpolar Health 2009, 68, 316–326. [Google Scholar]
- Philibert, A.; Schwartz, H.; Mergler, D. An exploratory study of diabetes in a First Nation community with respect to serum concentrations of p,p′-DDE and PCBs and fish consumption. Int. J. Environ. Res. Public Health 2009, 6, 3179–3189. [Google Scholar]
- Rylander, L.; Rignell-Hydbom, A.; Hagmar, L. A cross-sectional study of the association between persistent organochlorine pollutants and diabetes. Environ. Health 2005, 4, 1–6. [Google Scholar]
- Turyk, M.; Anderson, H.A.; Knobeloch, L.; Imm, P.; Persky, V.W. Prevalence of diabetes and body burdens of polychlorinated biphenyls, polybrominated diphenyl ethers, and p,p′-diphenyldichloroethene in Great Lakes sport fish consumers. Chemosphere 2009, 75, 674–679. [Google Scholar]
- Fujiyoshi, P.T.; Michalek, J.E.; Matsumura, F. Molecular epidemiologic evidence for diabetogenic effects of dioxin exposure in U.S. Air force veterans of the Vietnam war. Environ. Health Perspect 2006, 114, 1677–1683. [Google Scholar]
- Patel, C.J.; Bhattacharya, J.; Butte, A.J. An Environment-Wide Association study (EWAS) on type 2 diabetes mellitus. PLoS One 2010, 5, e10746. [Google Scholar]
- Higginbotham, G.R.; Huang, A.; Firestone, D.; Verrett, J.; Ress, J.; Campbell, A.D. Chemical and toxicological evaluations of isolated and synthetic chloro derivatives of dibenzo-p-dioxin. Nature 1968, 220, 702–703. [Google Scholar]
- Kimbrough, R.D. Toxicity of chlorinated hydrocarbons and related compounds. A review including chlorinated dibenzodioxins and chlorinated dibenzofurans. Arch. Environ. Health 1972, 25, 125–131. [Google Scholar]
- Schwetz, B.A.; Norris, J.M.; Sparschu, G.L.; Rowe, U.K.; Gehring, P.J.; Emerson, J.L.; Gerbig, C.G. Toxicology of chlorinated dibenzo-p-dioxins. Environ. Health Perspect 1973, 5, 87–99. [Google Scholar]
- Dragan, Y.P.; Schrenk, D. Animal studies addressing the carcinogenicity of TCDD (or related compounds) with an emphasis on tumour promotion. Food Addit. Contam 2000, 17, 289–302. [Google Scholar]
- Hernández, L.G.; van Steeg, H.; Luijten, M.; van Benthem, J. Mechanisms of non-genotoxic carcinogens and importance of a weight of evidence approach. Mutat. Res 2009, 682, 94–109. [Google Scholar]
- Fischer, B. Receptor-mediated effects of chlorinated hydrocarbons. Andrologia 2000, 32, 279–283. [Google Scholar]
- Yonemoto, J. The effects of dioxin on reproduction and development. Ind. Health 2000, 38, 259–268. [Google Scholar]
- Petersen, S.L.; Krishnan, S.; Hudgens, E.D. The aryl hydrocarbon receptor pathway and sexual differentiation of neuroendocrine functions. Endocrinology 2006, 147, S33–S42. [Google Scholar]
- McConnell, E.E.; Moore, J.A.; Dalgard, D.W. Toxicity of 2,3,7,8 tetrachlorodibenzo-p-dioxin in rhesus monkeys (Macaca mulatta) following a single oral dose. Toxicol. Appl. Pharmacol 1978, 43, 175–187. [Google Scholar]
- Olson, J.R.; Holscher, M.A.; Neal, R.A. Toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the golden Syrian hamster. Toxicol. Appl. Pharmacol 1980, 55, 67–78. [Google Scholar]
- Tuomisto, J.T.; Pohjanvirta, R.; Unkila, M.; Tuomisto, J. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced anorexia and wasting syndrome in rats: Aggravation after ventromedial hypothalamic lesion. Eur. J. Pharmacol 1995, 293, 309–317. [Google Scholar]
- Lindén, J.; Lensu, S.; Tuomisto, J.; Pohjanvirta, R. Dioxins, the aryl hydrocarbon receptor and the central regulation of energy balance. Front. Neuroendocrinol 2010, 31, 452–478. [Google Scholar]
- Seefeld, M.D.; Corbett, S.W.; Keesey, R.E.; Peterson, R.E. Characterization of the wasting syndrome in rats treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol 1984, 73, 311–322. [Google Scholar]
- Swift, L.L.; Gasiewicz, T.A.; Dunn, G.D.; Soulé, P.D.; Neal, R.A. Characterization of the hyperlipidemia in guinea pigs induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol 1981, 59, 489–499. [Google Scholar]
- Brewster, D.W.; Matsumura, F. TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin) reduces lipoprotein lipase activity in the adipose tissue of the guinea pig. Biochem. Biophys. Res. Commun 1984, 122, 810–817. [Google Scholar]
- Olsen, H.; Enan, E.; Matsumura, F. 2,3,7,8-Tetrachlorodibenzo-p-dioxin mechanism of action to reduce lipoprotein lipase activity in the 3T3-L1 preadipocyte cell line. J. Biochem. Mol. Toxicol 1998, 12, 29–39. [Google Scholar]
- Nishiumi, S.; Yabushita, Y.; Furuyashiki, T.; Fukuda, I.; Ashida, H. Involvement of SREBPs in 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced disruption of lipid metabolism in male guinea pig. Toxicol. Appl. Pharmacol 2008, 229, 281–289. [Google Scholar]
- Lo, R.; Celius, T.; Forgacs, A.L.; Dere, E.; MacPherson, L.; Harper, P.; Zacharewski, T.; Matthews, J. Identification of aryl hydrocarbon receptor binding targets in mouse hepatic tissue treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol 2011, 257, 38–47. [Google Scholar]
- Forgacs, A.L.; Kent, M.N.; Makley, M.K.; Mets, B.; DelRaso, N.; Jahns, G.L.; Burgoon, L.D.; Zacharewski, T.R.; Reo, N.V. Comparative metabolomic and genomic analyses of TCDD-elicited metabolic disruption in mouse and rat liver. Toxicol. Sci 2012, 125, 41–55. [Google Scholar]
- Angrish, M.M.; Dominici, C.Y.; Zacharewski, T.R. TCDD-elicited effects on liver, serum, and adipose lipid composition in C57BL/6 mice. Toxicol. Sci 2013, 131, 108–115. [Google Scholar]
- Ebner, K.; Brewster, D.W.; Matsumura, F. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on serum insulin and glucose levels in the rabbit. J. Environ. Sci. Health B 1988, 23, 427–438. [Google Scholar]
- Vogel, C.F.; Zhao, Y.; Wong, P.; Young, N.F.; Matsumura, F. The use of c-src knockout mice for the identification of the main toxic signaling pathway of TCDD to induce wasting syndrome. J. Biochem. Mol. Toxicol 2003, 17, 305–315. [Google Scholar]
- Kern, P.A.; Dicker-Brown, A.; Said, S.T.; Kennedy, R.; Fonseca, V.A. The stimulation of tumor necrosis factor and inhibition of glucose transport and lipoprotein lipase in adipose cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Metabolism 2002, 51, 65–68. [Google Scholar]
- Nishiumi, S.; Yoshida, M.; Azuma, T.; Yoshida, K.; Ashida, H. 2,3,7,8-tetrachlorodibenzo-p-dioxin impairs an insulin signaling pathway through the induction of tumor necrosis factor-alpha in adipocytes. Toxicol. Sci 2010, 115, 482–491. [Google Scholar]
- Li, W.; Vogel, C.F.; Matsumura, F. Studies on the cell treatment conditions to elicit lipolytic responses from 3T3-L1 adipocytes to TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Cell Biochem 2007, 102, 389–402. [Google Scholar]
- Fetissov, S.O.; Huang, P.; Zhang, Q.; Mimura, J.; Fujii-Kuriyama, Y.; Rannug, A.; Hökfelt, T.; Ceccatelli, S. Expression of hypothalamic neuropeptides after acute TCDD treatment and distribution of Ah receptor repressor. Regul. Pept 2004, 119, 113–124. [Google Scholar]
- Korkalainen, M.; Lindén, J.; Tuomisto, J.; Pohjanvirta, R. Effect of TCDD on mRNA expression of genes encoding bHLH/PAS proteins in rat hypothalamus. Toxicology 2005, 208, 1–11. [Google Scholar]
- Lindén, J.; Korkalainen, M.; Lensu, S.; Tuomisto, J.; Pohjanvirta, R. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and leptin on hypothalamic mRNA expression of factors participating in food intake regulation in a TCDD-sensitive and a TCDD-resistant rat strain. J. Biochem. Mol. Toxicol 2005, 19, 139–148. [Google Scholar]
- Lensu, S.; Miettinen, R.; Pohjanvirta, R.; Lindén, J.; Tuomisto, J. Assessment by c-Fos immunostaining of changes in brain neural activity induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and leptin in rats. Basic Clin. Pharmacol. Toxicol 2006, 98, 363–371. [Google Scholar]
- Moon, B.H.; Hong, C.G.; Kim, S.Y.; Kim, H.J.; Shin, S.K.; Kang, S.; Lee, K.J.; Kim, Y.K.; Lee, M.S.; Shin, K.H. A single administration of 2,3,7,8-tetrachlorodibenzo-p-dioxin that produces reduced food and water intake induces long-lasting expression of corticotropin-releasing factor, arginine vasopressin, and proopiomelanocortin in rat brain. Toxicol. Appl. Pharmacol 2008, 233, 314–322. [Google Scholar]
- Enan, E.; Liu, P.C.; Matsumura, F. 2,3,7,8-Tetrachlorodibenzo-p-dioxin causes reduction of glucose transporting activities in the plasma membranes of adipose tissue and pancreas from the guinea pig. J. Biol. Chem 1992, 267, 19785–19791. [Google Scholar]
- Enan, E.; Liu, P.C.; Matsumura, F. TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin) causes reduction in glucose uptake through glucose transporters on the plasma membrane of the guinea pig adipocyte. J. Environ. Sci. Health B 1992, 27, 495–510. [Google Scholar]
- Hsu, H.F.; Tsou, T.C.; Chao, H.R.; Kuo, Y.T.; Tsai, F.Y.; Yeh, S.C. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on adipogenic differentiation and insulin-induced glucose uptake in 3T3-L1 cells. J. Hazard. Mater 2010, 182, 649–655. [Google Scholar]
- Olsen, H.; Enan, E.; Matsumura, F. Regulation of glucose transport in the NIH 3T3 L1 preadipocyte cell line by TCDD. Environ. Health Perspect 1994, 102, 454–458. [Google Scholar]
- Enan, E.; Lasley, B.; Stewart, D.; Overstreet, J.; Vandevoort, C.A. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) modulates function of human luteinizing granulosa cells via cAMP signaling and early reduction of glucose transporting activity. Reprod. Toxicol 1996, 10, 191–198. [Google Scholar]
- Enan, E.; Matsumura, F. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)-induced changes in glucose transporting activity in guinea pigs, mice, and rats in vivo and in vitro. J. Biochem. Toxicol 1994, 9, 97–106. [Google Scholar]
- Liu, P.C.; Matsumura, F. Differential effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the “adipose-type” and “brain-type” glucose transporters in mice. Mol. Pharmacol 1995, 47, 65–73. [Google Scholar]
- Nagashima, H.; Matsumura, F. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)-induced down-regulation of glucose transporting activities in mouse 3T3-L1 preadipocyte. J. Environ. Sci. Health B 2002, 37, 1–14. [Google Scholar]
- Ishida, T.; Kan-o, S.; Mutoh, J.; Takeda, S.; Ishii, Y.; Hashiguchi, I.; Akamine, A.; Yamada, H. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced change in intestinal function and pathology: Evidence for the involvement of arylhydrocarbon receptor-mediated alteration of glucose transportation. Toxicol. Appl. Pharmacol 2005, 205, 89–97. [Google Scholar]
- Liu, P.C.; Matsumura, F. TCDD suppresses insulin-responsive glucose transporter (GLUT-4) gene expression through C/EBP nuclear transcription factors in 3T3-L1 adipocytes. J. Biochem. Mol. Toxicol 2006, 20, 79–87. [Google Scholar]
- Tonack, S.; Kind, K.; Thompson, J.G.; Wobus, A.M.; Fischer, B.; Santos, A.N. Dioxin affects glucose transport via the arylhydrocarbon receptor signal cascade in pluripotent embryonic carcinoma cells. Endocrinology 2007, 148, 5902–5912. [Google Scholar]
- Matsumura, F. Mechanism of action of dioxin-type chemicals, pesticides, and other xenobiotics affecting nutritional indexes. Am. J. Clin. Nutr 1995, 61, 695S–701S. [Google Scholar]
- Gorski, J.R.; Rozman, K. Dose-response and time course of hypothyroxinemia and hypoinsulinemia and characterization of insulin hypersensitivity in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-treated rats. Toxicology 1987, 44, 297–307. [Google Scholar]
- Gorski, J.R.; Muzi, G.; Weber, L.W.; Pereira, D.W.; Arceo, R.J.; Iatropoulos, M.J.; Rozman, K. Some endocrine and morphological aspects of the acute toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol. Pathol 1988, 16, 313–320. [Google Scholar]
- Novelli, M.; Piaggi, S.; de Tata, V. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced impairment of glucose-stimulated insulin secretion in isolated rat pancreatic islets. Toxicol. Lett 2005, 156, 307–314. [Google Scholar]
- Kurita, H.; Yoshioka, W.; Nishimura, N.; Kubota, N.; Kadowaki, T.; Tohyama, C. Aryl hydrocarbon receptor-mediated effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on glucose-stimulated insulin secretion in mice. J. Appl. Toxicol 2009, 29, 689–694. [Google Scholar]
- Panten, U.; Kriegstein, E.; Poser, W.; Schönborn, J.; Hasselblatt, A. Effects of l-leucine and alpha-ketoisocaproic acid upon insulin secretion and metabolism of isolated pancreatic islets. FEBS Lett 1972, 20, 225–228. [Google Scholar]
- Lenzen, S.; Formanek, H.; Panten, U. Signal function of metabolism of neutral amino acids and 2-keto acids for initiation of insulin secretion. J. Biol. Chem 1982, 257, 6631–6633. [Google Scholar]
- Lembert, N.; Idahl, L.A. Alpha-ketoisocaproate is not a true substrate for ATP production by pancreatic beta-cell mitochondria. Diabetes 1998, 47, 339–344. [Google Scholar]
- Matschinsky, F.M. Glucokinase as glucose sensor and metabolic signal generator in pancreatic beta-cells and hepatocytes. Diabetes 1990, 39, 647–652. [Google Scholar]
- Malaisse, W.J.; Malaisse-Lagae, F.; Rasschaert, J.; Zähner, D.; Sener, A.; Davies, D.R.; van Schaftingen, E. The fuel concept for insulin release: Regulation of glucose phosphorylation in pancreatic islets. Biochem. Soc. Trans 1990, 18, 107–108. [Google Scholar]
- Efrat, S.; Tal, M.; Lodish, H.F. The pancreatic beta-cell glucose sensor. Trends Biochem. Sci 1994, 19, 535–538. [Google Scholar]
- Orci, L.; Ravazzola, M.; Baetens, D.; Inman, L.; Amherdt, M.; Peterson, R.G.; Newgard, C.B.; Johnson, J.H.; Unger, R.H. Evidence that down-regulation of beta-cell glucose transporters in non-insulin-dependent diabetes may be the cause of diabetic hyperglycemia. Proc. Natl. Acad. Sci. USA 1990, 87, 9953–9957. [Google Scholar]
- Thorens, B.; Weir, G.C.; Leahy, J.L.; Lodish, H.F.; Bonner-Weir, S. Reduced expression of the liver/beta-cell glucose transporter isoform in glucose-insensitive pancreatic beta cells of diabetic rats. Proc. Natl. Acad. Sci. USA 1990, 87, 6492–6496. [Google Scholar]
- Valera, A.; Solanes, G.; Fernández-Alvarez, J.; Pujol, A.; Ferrer, J.; Asins, G.; Gomis, R.; Bosch, F. Expression of GLUT-2 antisense RNA in beta cells of transgenic mice leads to diabetes. J. Biol. Chem 1994, 269, 28543–28546. [Google Scholar]
- Fisher, J.M.; Jones, K.W.; Whitlock, J.P., Jr. Activation of transcription as a general mechanism of 2,3,7,8-tetrachlorodibenzo-p-dioxin action. Mol. Carcinog 1989, 1, 216–221. [Google Scholar]
- Silbergeld, E.K.; Gasiewicz, T.A. Dioxins and the Ah receptor. Am. J. Ind. Med 1989, 16, 455–474. [Google Scholar]
- Bock, K.W.; Köhle, C. Ah receptor: Dioxin-mediated toxic responses as hints to deregulated physiologic functions. Biochem. Pharmacol 2006, 72, 393–404. [Google Scholar]
- Hankinson, O. The aryl hydrocarbon receptor complex. Annu. Rev. Pharmacol. Toxicol 1995, 35, 307–340. [Google Scholar]
- Gorski, J.R.; Weber, L.W.; Rozman, K. Tissue-specific alterations of de novo fatty acid synthesis in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-treated rats. Arch. Toxicol 1988, 62, 146–151. [Google Scholar]
- Weber, L.W.; Lebofsky, M.; Greim, H.; Rozman, K. Key enzymes of gluconeogenesis are dose-dependently reduced in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-treated rats. Arch. Toxicol 1991, 65, 119–123. [Google Scholar]
- Weber, L.W.; Lebofsky, M.; Stahl, B.U.; Kettrup, A.; Rozman, K. Comparative toxicity of four chlorinated dibenzo-p-dioxins (CDDs) and their mixture. Part II: Structure-activity relationships with inhibition of hepatic phosphoenolpyruvate carboxykinase, pyruvate carboxylase, and gamma-glutamyl transpeptidase activities. Arch. Toxicol 1992, 66, 478–483. [Google Scholar]
- Stahl, B.U.; Beer, D.G.; Weber, L.W.; Rozman, K. Reduction of hepatic phosphoenolpyruvate carboxykinase (PEPCK) activity by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is due to decreased mRNA levels. Toxicology 1993, 79, 81–95. [Google Scholar]
- Fan, F.; Yan, B.; Wood, G.; Viluksela, M.; Rozman, K.K. Cytokines (IL-1beta and TNFalpha) in relation to biochemical and immunological effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in rats. Toxicology 1997, 116, 9–16. [Google Scholar]
- Croutch, C.R.; Lebofsky, M.; Schramm, K.W.; Terranova, P.F.; Rozman, K.K. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) and 1,2,3,4,7,8-hexachlorodibenzo-p-dioxin (HxCDD) alter body weight by decreasing insulin-like growth factor I (IGF-I) signaling. Toxicol. Sci 2005, 85, 560–571. [Google Scholar]
- Fletcher, N.; Wahlström, D.; Lundberg, R.; Nilsson, C.B.; Nilsson, K.C.; Stockling, K.; Hellmold, H.; Håkansson, H. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) alters the mRNA expression of critical genes associated with cholesterol metabolism, bile acid biosynthesis, and bile transport in rat liver: a microarray study. Toxicol. Appl. Pharmacol 2005, 207, 1–24. [Google Scholar]
- Sato, S.; Shirakawa, H.; Tomita, S.; Ohsaki, Y.; Haketa, K.; Tooi, O.; Santo, N.; Tohkin, M.; Furukawa, Y.; Gonzalez, F.J.; et al. Low-dose dioxins alter gene expression related to cholesterol biosynthesis, lipogenesis, and glucose metabolism through the aryl hydrocarbon receptor-mediated pathway in mouse liver. Toxicol. Appl. Pharmacol 2008, 229, 10–19. [Google Scholar]
- Zhang, W.; Sargis, R.M.; Volden, P.A.; Carmean, C.M.; Sun, X.J.; Brady, M.J. PCB 126 and other dioxin-like PCBs specifically suppress hepatic PEPCK expression via the aryl hydrocarbon receptor. PLoS One 2012, 7, e37103. [Google Scholar]
- Puga, A.; Maier, A.; Medvedovic, M. The transcriptional signature of dioxin in human hepatoma HepG2 cells. Biochem. Pharmacol 2000, 60, 1129–1142. [Google Scholar]
- Diani-Moore, S.; Ram, P.; Li, X.; Mondal, P.; Youn, D.Y.; Sauve, A.A.; Rifkind, A.B. Identification of the aryl hydrocarbon receptor target gene tiparp as a mediator of suppression of hepatic gluconeogenesis by 2,3,7,8-tetrachlorodibenzo-p-dioxin and of nicotinamide as a corrective agent for this effect. J. Biol. Chem 2010, 285, 38801–38810. [Google Scholar]
- Donath, M.Y.; Böni-Schnetzler, M.; Ellingsgaard, H.; Halban, P.A.; Ehses, J.A. Cytokine production by islets in health and diabetes: cellular origin, regulation and function. Trends Endocrinol. Metab 2010, 21, 261–267. [Google Scholar]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar]
- Donath, M.Y.; Schumann, D.M.; Faulenbach, M.; Ellingsgaard, H.; Perren, A.; Ehses, J.A. Islet inflammation in type 2 diabetes: From metabolic stress to therapy. Diabetes Care 2008, 31, S161–S164. [Google Scholar]
- Matsumura, F. On the significance of the role of cellular stress response reactions in the toxic actions of dioxin. Biochem. Pharmacol 2003, 66, 527–540. [Google Scholar]
- Matsumura, F.; Vogel, C.F. Evidence supporting the hypothesis that one of the main functions of the aryl hydrocarbon receptor is mediation of cell stress responses. Biol. Chem 2006, 387, 1189–1194. [Google Scholar]
- Kim, M.J.; Pelloux, V.; Guyot, E.; Tordjman, J.; Bui, L.C.; Chevallier, A.; Forest, C.; Benelli, C.; Clément, K.; Barouki, R. Inflammatory pathway genes belong to major targets of persistent organic pollutants in adipose cells. Environ. Health Perspect 2012, 120, 508–514. [Google Scholar]
- Vogel, C.F.; Kahn, E.M.; Leung, P.S.; Gershwin, M.E.; Chang, W.L.; Wu, D.; Haarmann-Stemmann, T.; Hoffmann, A.; Denison, M.S. Cross-talk between Aryl Hydrocarbon Receptor and the inflammatory response: A Role for NF-κB. J. Biol. Chem 2014, 289, 1866–1875. [Google Scholar]
- Dong, B.; Matsumura, F. Roles of cytosolic phospholipase A2 and Src kinase in the early action of 2,3,7,8-tetrachlorodibenzo-p-dioxin through a nongenomic pathway in MCF10A cells. Mol. Pharmacol 2008, 74, 255–263. [Google Scholar]
- Dong, B.; Matsumura, F. The conversion of rapid TCCD nongenomic signals to persistent inflammatory effects via select protein kinases in MCF10A cells. Mol. Endocrinol 2009, 23, 549–558. [Google Scholar]
- Li, W.; Matsumura, F. Significance of the nongenomic, inflammatory pathway in mediating the toxic action of TCDD to induce rapid and long-term cellular responses in 3T3-L1 adipocytes. Biochemistry 2008, 47, 13997–14008. [Google Scholar]
- Sciullo, E.M.; Dong, B.; Vogel, C.F.; Matsumura, F. Characterization of the pattern of the nongenomic signaling pathway through which TCDD-induces early inflammatory responses in U937 human macrophages. Chemosphere 2009, 74, 1531–1537. [Google Scholar]
- Dong, B.; Nishimura, N.; Vogel, C.F.; Tohyama, C.; Matsumura, F. TCDD-induced cyclooxygenase-2 expression is mediated by the nongenomic pathway in mouse MMDD1 macula densa cells and kidneys. Biochem. Pharmacol 2010, 79, 487–497. [Google Scholar]
- Xu, G.; Li, Y.; Yoshimoto, K.; Chen, G.; Wan, C.; Iwata, T.; Mizusawa, N.; Duan, Z.; Liu, J.; Jiang, J. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced inflammatory activation is mediated by intracellular free calcium in microglial cells. Toxicology 2013, 308, 158–167. [Google Scholar]
- Matsumura, F. The significance of the nongenomic pathway in mediating inflammatory signaling of the dioxin-activated Ah receptor to cause toxic effects. Biochem. Pharmacol 2009, 77, 608–626. [Google Scholar]
- Prentki, M.; Nolan, C.J. Islet beta cell failure in type 2 diabetes. J. Clin. Investig 2006, 116, 1802–1812. [Google Scholar]
- Ashcroft, F.M.; Rorsman, P. Diabetes mellitus and the β cell: The last ten years. Cell 2012, 148, 1160–1171. [Google Scholar]
- Hectors, T.L.; Vanparys, C.; van der Ven, K.; Martens, G.A.; Jorens, P.G.; van Gaal, L.F.; Covaci, A.; de Coen, W.; Blust, R. Environmental pollutants and type 2 diabetes: A review of mechanisms that can disrupt beta cell function. Diabetologia 2011, 54, 1273–1290. [Google Scholar]
- Asfari, M.; Janjic, D.; Meda, P.; Li, G.; Halban, P.A.; Wollheim, C.B. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology 1992, 130, 167–178. [Google Scholar]
- Merglen, A.; Theander, S.; Rubi, B.; Chaffard, G.; Wollheim, C.B.; Maechler, P. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 2004, 145, 667–678. [Google Scholar]
- Piaggi, S.; Novelli, M.; Martino, L.; Masini, M.; Raggi, C.; Orciuolo, E.; Masiello, P.; Casini, A.; de Tata, V. Cell death and impairment of glucose-stimulated insulin secretion induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the beta-cell line INS-1E. Toxicol. Appl. Pharmacol 2007, 220, 333–340. [Google Scholar]
- Mitrou, P.I.; Dimitriadis, G.; Raptis, S.A. Toxic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds. Eur. J. Intern. Med 2001, 12, 406–411. [Google Scholar]
- Sakurai, K.; Katoh, M.; Someno, K.; Fujimoto, Y. Apoptosis and mitochondrial damage in INS-1 cells treated with alloxan. Biol. Pharm. Bull 2001, 24, 876–882. [Google Scholar]
- Hanneman, W.H.; Legare, M.E.; Barhoumi, R.; Burghardt, R.C.; Safe, S.; Tiffany-Castiglioni, E. Stimulation of calcium uptake in cultured rat hippocampal neurons by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology 1996, 112, 19–28. [Google Scholar]
- Puga, A.; Hoffer, A.; Zhou, S.; Bohm, J.M.; Leikauf, G.D.; Shertzer, H.G. Sustained increase in intracellular free calcium and activation of cyclooxigenase-2 expression in mouse hepatoma cells treated with dioxin. Biochem. Pharmacol 1997, 54, 1287–1296. [Google Scholar]
- Tannheimer, S.L.; Barton, S.L.; Ethier, S.P.; Burchiel, S.W. Carcinogenic polycyclic aromatic hydrocarbons increase intracellular Ca2+ and cell proliferation in primary human mammary epithelial cells. Carcinogenesis 1997, 18, 1177–1182. [Google Scholar]
- N’Diaye, M.; le Ferrec, E.; Lagadic-Gossmann, D.; Corre, S.; Gilot, D.; Lecureur, V.; Monteiro, P.; Rauch, C.; Galibert, M.D.; Fardel, O. Aryl hydrocarbon receptor- and calcium-dependent induction of the chemokine CCL1 by the environmental contaminant benzo[a]pyrene. J. Biol. Chem 2006, 281, 19906–19915. [Google Scholar]
- Dale, Y.R.; Eltom, S.E. Calpain mediates the dioxin-induced activation and down-regulation of the aryl hydrocarbon receptor. Mol. Pharmacol 2006, 70, 1481–1487. [Google Scholar]
- Xie, A.; Walker, N.J.; Wang, D. Dioxin (2,3,7,8-tetrachlorodibenzo-p-dioxin) enhances triggered afterdepolarizations in rat ventricular myocytes. Cardiovasc. Toxicol 2006, 6, 99–110. [Google Scholar]
- Kim, S.Y.; Lee, H.G.; Choi, E.J.; Park, K.Y.; Yang, J.H. TCDD alters PKC signaling pathways in developing neuronal cells in culture. Chemosphere 2007, 67, S421–S427. [Google Scholar]
- Monteiro, P.; Gilot, D.; le Ferrec, E.; Rauch, C.; Lagadic-Gossmann, D.; Fardel, O. Dioxin-mediated up-regulation of aryl hydrocarbon receptor target genes is dependent on the calcium/calmodulin/CaMKIalpha pathway. Mol. Pharmacol 2008, 73, 769–777. [Google Scholar]
- Sul, D.; Kim, H.S.; Cho, E.K.; Lee, M.; Kim, H.S.; Jung, W.W.; Hwang, K.W.; Park, S.Y. 2,3,7,8-TCDD neurotoxicity in neuroblastoma cells is caused by increased oxidative stress, intracellular calcium levels, and tau phosphorylation. Toxicology 2009, 255, 65–71. [Google Scholar]
- Kobayashi, D.; Ahmed, S.; Ishida, M.; Kasai, S.; Kikuchi, H. Calcium/calmodulin signaling elicits release of cytochrome c during 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced apoptosis in the human lymphoblastic T-cell line, L-MAT. Toxicology 2009, 258, 25–32. [Google Scholar]
- Kim, Y.H.; Shim, Y.J.; Shin, Y.J.; Sul, D.; Lee, E.; Min, B.H. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induces calcium influx through T-type calcium channel and enhances lysosomal exocytosis and insulin secretion in INS-1 cells. Int. J. Toxicol 2009, 28, 151–161. [Google Scholar]
- Morales-Hernández, A.; Sánchez-Martín, F.J.; Hortigón-Vinagre, M.P.; Henao, F.; Merino, J.M. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces apoptosis by disruption of intracellular calcium homeostasis in human neuronal cell line SHSY5Y. Apoptosis 2012, 17, 1170–1181. [Google Scholar]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev 2005, 85, 757–810. [Google Scholar]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol 2003, 4, 552–565. [Google Scholar]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S.; Calcium, A.T.P. ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol 2004, 287, C817–C833. [Google Scholar]
- Senft, A.P.; Dalton, T.P.; Nebert, D.W.; Genter, M.B.; Hutchinson, R.J.; Shertzer, H.G. Dioxin increases reactive oxygen production in mouse liver mitochondria. Toxicol. Appl. Pharmacol 2002, 185, 74–75. [Google Scholar]
- Shen, D.; Dalton, T.P.; Nerbert, D.W.; Shertzer, H.G. Glutathione redox state regulates mitochondrial reactive oxygen production. J. Biol. Chem 2005, 280, 25305–25312. [Google Scholar]
- Shertzer, H.G.; Genter, M.B.; Shen, D.; Nebert, D.W.; Chen, Y.; Dalton, T.P. TCDD decreases ATP levels and increases reactive oxygen production through changes in mitochondrial F0F1-ATP synthase and ubiquinone. Toxicol. Appl. Pharmacol 2006, 217, 363–374. [Google Scholar]
- Maechler, P.; Carobbio, S.; Rubi, B. In beta-cells, mitochondria integrate and generate metabolic signals controlling insulin secretion. Int. J. Biochem. Cell Biol 2006, 38, 696–709. [Google Scholar]
- Martino, L.; Novelli, M.; Masini, M.; Chimenti, D.; Piaggi, S.; Masiello, P.; de Tata, V. Dehydroascorbate protection against dioxin-induced toxicity in the beta-cell line INS-1E. Toxicol. Lett 2009, 189, 27–34. [Google Scholar]
- Martino, L.; Masini, M.; Novelli, M.; Giacopelli, D.; Beffy, P.; Masiello, P.; de Tata, V. The aryl receptor inhibitor epigallocatechin-3-gallate protects INS-1E beta-cell line against acute dioxin toxicity. Chemosphere 2013, 93, 1447–1455. [Google Scholar]
- Mannella, C.A. The relevance of mitochondrial membrane topology to mitochondrial function. Biochim. Biophys. Acta 2006, 1762, 140–147. [Google Scholar]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev 2007, 87, 99–163. [Google Scholar]
- Zhang, Z.; Ding, Y.; Dai, X.; Wang, J.; Li, Y. Epigallocatechin-3-gallate protects pro-inflammatory cytokine induced injuries in insulin-producing cells through the mitochondrial pathway. Eur. J. Pharmacol 2011, 670, 311–316. [Google Scholar]
- Lim, S.; Cho, Y.M.; Park, K.S.; Lee, H.K. Persistent organic pollutants, mitochondrial dysfunction, and metabolic syndrome. Ann. N. Y. Acad. Sci 2010, 1201, 166–176. [Google Scholar]
- Lee, H.K. Mitochondrial dysfunction and insulin resistance: The contribution of dioxin-like substances. Diabetes Metab. J 2011, 35, 207–215. [Google Scholar]
- Park, W.H.; Jun, D.W.; Kim, J.T.; Jeong, J.H.; Park, H.; Chang, Y.S.; Park, K.S.; Lee, H.K.; Pak, Y.K. Novel cell-based assay reveals associations of circulating serum AhR-ligands with metabolic syndrome and mitochondrial dysfunction. Biofactors 2013, 39, 494–504. [Google Scholar]
- Kennedy, L.H.; Sutter, C.H.; Leon Carrion, S.; Tran, Q.T.; Bodreddigari, S.; Kensicki, E.; Mohney, R.P.; Sutter, T.R. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated production of reactive oxygen species is an essential step in the mechanism of action to accelerate human keratinocyte differentiation. Toxicol. Sci 2013, 132, 235–249. [Google Scholar]
- Pereira, S.P.; Pereira, G.C.; Pereira, C.V.; Carvalho, F.S.; Cordeiro, M.H.; Mota, P.C.; Ramalho-Santos, J.; Moreno, A.J.; Oliveira, P.J. Dioxin-induced acute cardiac mitochondrial oxidative damage and increased activity of ATP-sensitive potassium channels in Wistar rats. Environ. Pollut 2013, 180, 281–290. [Google Scholar] [Green Version]
- Fröjdö, S.; Vidal, H.; Pirola, L. Alterations of insulin signaling in type 2 diabetes: A review of the current evidence from humans. Biochim. Biophys. Acta 2009, 1792, 83–92. [Google Scholar]
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar]
- Abdul-Ghani, M.A.; DeFronzo, R.A. Mitochondrial dysfunction, insulin resistance, and type 2 diabetes mellitus. Curr. Diabetes Rep 2008, 8, 173–178. [Google Scholar]
- Cheng, Z.; Tseng, Y.; White, M.F. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol. Metab 2010, 21, 589–598. [Google Scholar]
- López-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-García, C.; Valcárcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar]
- Ruzzin, J.; Petersen, R.; Meugnier, E.; Madsen, L.; Lock, E.J.; Lillefosse, H.; Ma, T.; Pesenti, S.; Sonne, S.B.; Marstrand, T.T.; et al. Persistent organic pollutant exposure leads to insulin resistance syndrome. Environ. Health Perspect 2010, 118, 465–471. [Google Scholar]
- Slezak, B.P.; Hatch, G.E.; DeVito, M.J.; Diliberto, J.J.; Slade, R.; Crissman, K.; Hassoun, E.; Birnbaum, L.S. Oxidative stress in female B6C3F1 mice following acute and subchronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol. Sci 2000, 54, 390–398. [Google Scholar]
- Regnier, S.M.; Sargis, R.M. Adipocytes under assault: Environmental disruption of adipose physiology. Biochim. Biophys. Acta 2014, 1842, 520–533. [Google Scholar]
- Baillie-Hamilton, P.F. Chemical toxins: A hypothesis to explain the global obesity epidemic. J. Altern. Complement. Med 2002, 8, 185–192. [Google Scholar]
- Arsenescu, V.; Arsenescu, R.I.; King, V.; Swanson, H.; Cassis, L.A. Polychlorinated biphenyl-77 induces adipocyte differentiation and proinflammatory adipokines and promotes obesity and atherosclerosis. Environ. Health Perspect 2008, 116, 761–768. [Google Scholar]
- Ibrahim, M.M.; Fjære, E.; Lock, E.J.; Naville, D.; Amlund, H.; Meugnier, E.; Frøyland, L.; Le Magueresse Battistoni, B.; Madsen, L.; Jessen, N.; et al. Chronic consumption of farmed salmon containing persistent organic pollutants causes insulin resistance and obesity in mice. PLoS One 2011, 6, e25170. [Google Scholar]
- Gauthier, M.S.; Rabasa-Lhoret, R.; Prud’homme, D.; Karelis, A.D.; Geng, D.; van Bavel, B.; Ruzzin, J. The metabolically healthy but obese phenotype is associated with lower plasma levels of persistent organic pollutants as compared to the metabolically abnormal obese phenotype. J. Clin. Endocrinol. Metab 2014. [Google Scholar] [CrossRef]
- Tan, Z.; Chang, X.; Puga, A.; Xia, Y. Activation of mitogen-activated protein kinases (MAPKs) by aromatic hydrocarbons: Role in the regulation of aryl hydrocarbon receptor (AHR) function. Biochem. Pharmacol 2002, 64, 771–780. [Google Scholar]
- Kwon, M.J.; Jeong, K.S.; Choi, E.J.; Lee, B.H. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)-induced activation of mitogen-activated protein kinase signaling pathway in Jurkat T cells. Pharmacol. Toxicol 2003, 93, 186–190. [Google Scholar]
- Weiss, C.; Faust, D.; Dürk, H.; Kolluri, S.K.; Pelzer, A.; Schneider, S.; Dietrich, C.; Oesch, F.; Göttlicher, M. TCDD induces c-jun expression via a novel Ah (dioxin) receptor-mediated p38-MAPK-dependent pathway. Oncogene 2005, 24, 4975–4983. [Google Scholar]
- Park, S.J.; Yoon, W.K.; Kim, H.J.; Son, H.Y.; Cho, S.W.; Jeong, K.S.; Kim, T.H.; Kim, S.H.; Kim, S.R.; Ryu, S.Y. 2,3,7,8-Tetrachlorodibenzo-p-dioxin activates ERK and p38 mitogen-activated protein kinases in RAW 264.7 cells. Anticancer Res 2005, 25, 2831–2836. [Google Scholar]
- Sciullo, E.M.; Vogel, C.F.; Wu, D.; Murakami, A.; Ohigashi, H.; Matsumura, F. Effects of selected food phytochemicals in reducing the toxic actions of TCDD and p,p′-DDT in U937 macrophages. Arch. Toxicol 2010, 84, 957–966. [Google Scholar]
- Mukai, R.; Shirai, Y.; Saito, N.; Fukuda, I.; Nishiumi, S.; Yoshida, K.; Ashida, H. Suppression mechanisms of flavonoids on aryl hydrocarbon receptor-mediated signal transduction. Arch. Biochem. Biophys 2010, 501, 134–141. [Google Scholar]
- Palermo, C.M.; Westlake, C.A.; Gasiewicz, T.A. Epigallocatechin gallate inhibits aryl hydrocarbon receptor gene transcription through an indirect mechanism involving binding to a 90 kDa heat shock protein. Biochemistry 2005, 44, 5041–5052. [Google Scholar]
- Park, S.; Dong, B.; Matsumura, F. Rapid activation of c-Src kinase by dioxin is mediated by the Cdc37-HSP90 complex as part of Ah receptor signaling in MCF10A cells. Biochemistry 2007, 46, 899–908. [Google Scholar]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Aqostinis, P.; Aquirre-Ghiso, J.A.; Ait-Mohamed, O.; Ait-Si-Ali, S.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar]
- Fiorito, F.; Ciarcia, R.; Granato, G.E.; Marfe, G.; Iovane, V.; Florio, S.; de Martino, L.; Pagnini, U. 2,3,7,8-tetrachlorodibenzo-p-dioxin induced autophagy in a bovine kidney cell line. Toxicology 2011, 290, 258–270. [Google Scholar]
- Masini, M.; Bugliani, M.; Lupi, R.; del Guerra, S.; Boggi, U.; Filipponi, F.; Marselli, L.; Masiello, P.; Marchetti, P. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 2009, 52, 1083–1086. [Google Scholar]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 2008, 8, 325–332. [Google Scholar]
- Jung, H.S.; Chung, K.W.; Won Kim, J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.H.; Kim, J.W.; et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab 2008, 8, 318–324. [Google Scholar]
- Meijer, A.J.; Codogno, P. Autophagy: A sweet process in diabetes. Cell Metab 2008, 8, 275–276. [Google Scholar]
- Fujitani, Y.; Ueno, T.; Watada, H. Autophagy in health and disease. 4. The role of pancreatic beta-cell autophagy in health and diabetes. Am. J. Physiol. Cell Physiol 2010, 299, C1–C6. [Google Scholar]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar]
- Martino, L.; Masini, M.; Novelli, M.; Beffy, P.; Bugliani, M.; Marselli, L.; Masiello, P.; Marchetti, P.; de Tata, V. Palmitate activates autophagy in INS-1E β-cells and in isolated rat and human pancreatic islets. PLoS One 2012, 7, e36188. [Google Scholar]

© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Tata, V. Association of Dioxin and Other Persistent Organic Pollutants (POPs) with Diabetes: Epidemiological Evidence and New Mechanisms of Beta Cell Dysfunction. Int. J. Mol. Sci. 2014, 15, 7787-7811. https://doi.org/10.3390/ijms15057787
De Tata V. Association of Dioxin and Other Persistent Organic Pollutants (POPs) with Diabetes: Epidemiological Evidence and New Mechanisms of Beta Cell Dysfunction. International Journal of Molecular Sciences. 2014; 15(5):7787-7811. https://doi.org/10.3390/ijms15057787
Chicago/Turabian StyleDe Tata, Vincenzo. 2014. "Association of Dioxin and Other Persistent Organic Pollutants (POPs) with Diabetes: Epidemiological Evidence and New Mechanisms of Beta Cell Dysfunction" International Journal of Molecular Sciences 15, no. 5: 7787-7811. https://doi.org/10.3390/ijms15057787
APA StyleDe Tata, V. (2014). Association of Dioxin and Other Persistent Organic Pollutants (POPs) with Diabetes: Epidemiological Evidence and New Mechanisms of Beta Cell Dysfunction. International Journal of Molecular Sciences, 15(5), 7787-7811. https://doi.org/10.3390/ijms15057787