Cellular Levels of 8-Oxoguanine in either DNA or the Nucleotide Pool Play Pivotal Roles in Carcinogenesis and Survival of Cancer Cells

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

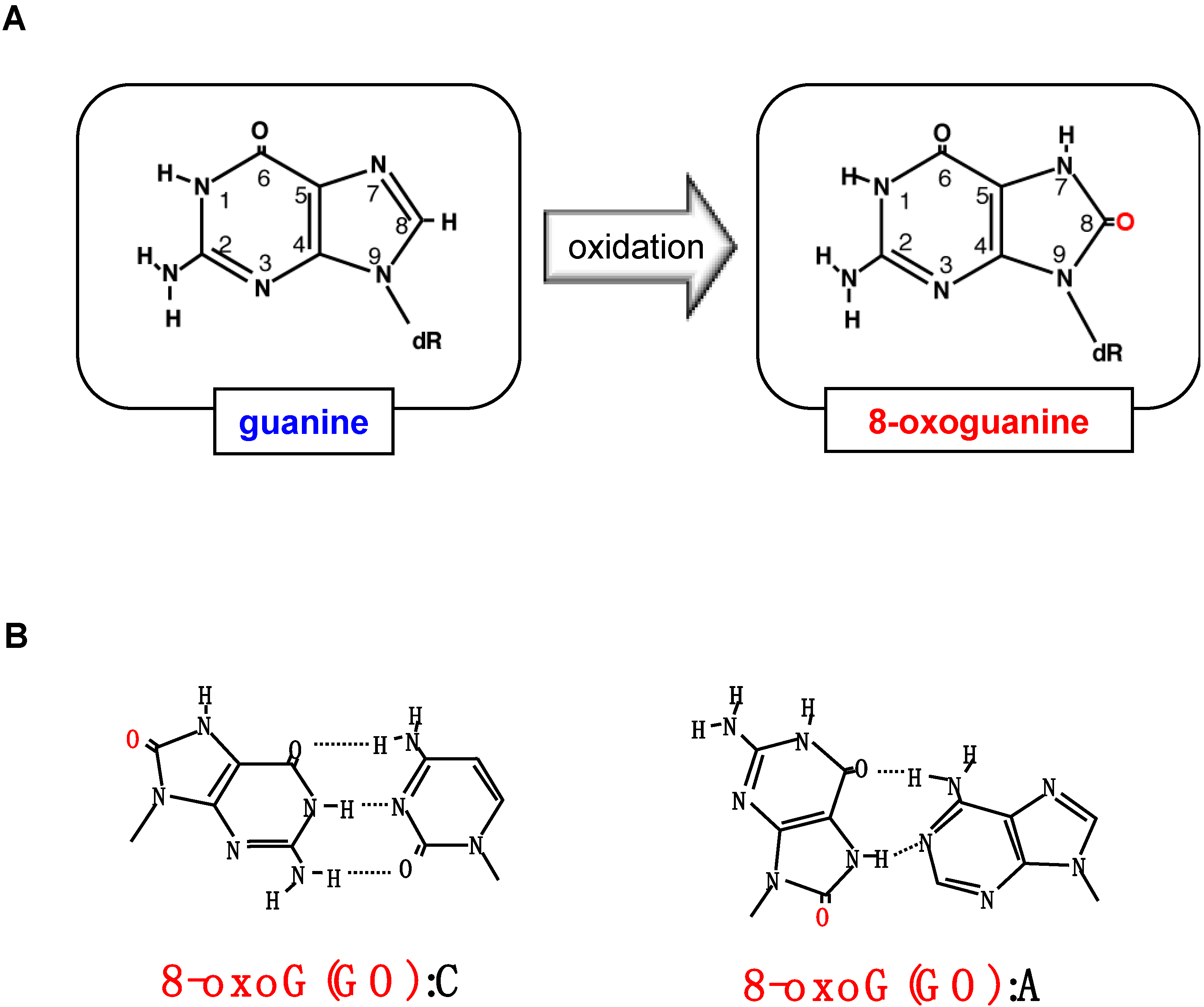
2. Formation of 8-Oxoguanine in Nucleic Acids and Its Origin and Distribution in DNA
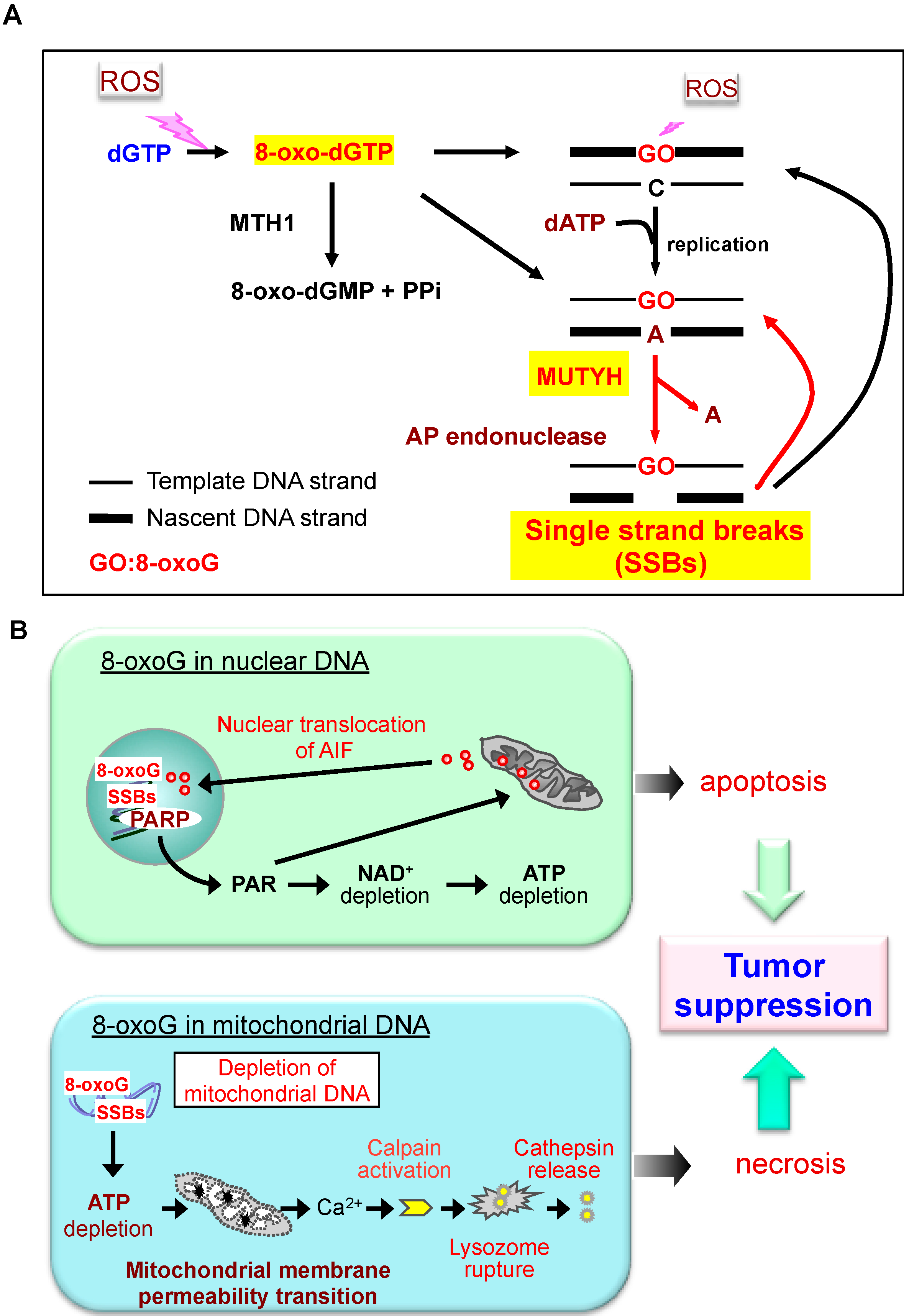
3. Defense Systems to Minimize Accumulation of 8-oxoG in Mammalian DNA

4. Altered Susceptibility to Spontaneous Mutagenesis and Carcinogenesis in Mice Deficient in 8-oxoG Defense Systems
5. Altered Susceptibility to Induced Tumorigenesis in Mice Deficient in the Defense Systems
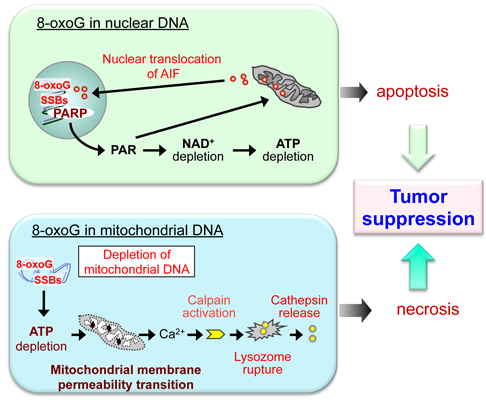
6. Do the Defense Systems that Minimize 8-oxoG Accumulation in DNA Contribute to the Survival of Cancer Cells?

7. Future Perspectives
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kang, D.; Takeshige, K.; Sekiguchi, M.; Singh, K.K. Introduction. In Mitochondrial DNA Mutations in Aging, Disease and Cancer; Singh, K.K., Ed.; Springer/Verlag Berlin: Heidelberg, Germany; New York, NY, USA, 1998; pp. 1–15. [Google Scholar]
- Ames, B.N. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef]
- Kasai, H.; Nishimura, S. Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res. 1984, 12, 2137–2145. [Google Scholar] [CrossRef]
- Maki, H. Origins of spontaneous mutations: Specificity and directionality of base-substitution, frameshift, and sequence-substitution mutageneses. Annu. Rev. Genet. 2002, 36, 279–303. [Google Scholar] [CrossRef]
- Nakabeppu, Y.; Sakumi, K.; Sakamoto, K.; Tsuchimoto, D.; Tsuzuki, T.; Nakatsu, Y. Mutagenesis and carcinogenesis caused by the oxidation of nucleic acids. Biol. Chem. 2006, 387, 373–379. [Google Scholar]
- Oka, S.; Nakabeppu, Y. DNA glycosylase encoded by MUTYH functions as a molecular switch for programmed cell death under oxidative stress to suppress tumorigenesis. Cancer Sci. 2011, 102, 677–682. [Google Scholar] [CrossRef]
- Mo, J.; Maki, H.; Sekiguchi, M. Hydrolytic elimination of a mutagenic nucleotide, 8-oxodGTP, by human 18-kilodalton protein: sanitization of nucleotide pool. Proc. Natl. Acad. Sci. USA 1992, 89, 11021–11025. [Google Scholar] [CrossRef]
- Kamiya, H.; Kasai, H. Formation of 2-hydroxydeoxyadenosine triphosphate, an oxidatively damaged nucleotide, and its incorporation by DNA polymerases. Steady-state kinetics of the incorporation. J. Biol. Chem. 1995, 270, 19446–19450. [Google Scholar] [CrossRef]
- Pursell, Z.F.; McDonald, J.T.; Mathews, C.K.; Kunkel, T.A. Trace amounts of 8-oxo-dGTP in mitochondrial dNTP pools reduce DNA polymerase gamma replication fidelity. Nucleic Acids Res. 2008, 36, 2174–2181. [Google Scholar] [CrossRef]
- Maki, H.; Sekiguchi, M. MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature 1992, 355, 273–275. [Google Scholar] [CrossRef]
- Katafuchi, A.; Nohmi, T. DNA polymerases involved in the incorporation of oxidized nucleotides into DNA: Their efficiency and template base preference. Mutat. Res. 2010, 703, 24–31. [Google Scholar] [CrossRef]
- Ohno, M.; Miura, T.; Furuichi, M.; Tominaga, Y.; Tsuchimoto, D.; Sakumi, K.; Nakabeppu, Y. A genome-wide distribution of 8-oxoguanine correlates with the preferred regions for recombination and single nucleotide polymorphism in the human genome. Genome Res. 2006, 16, 567–575. [Google Scholar] [CrossRef]
- Sakai, Y.; Furuichi, M.; Takahashi, M.; Mishima, M.; Iwai, S.; Shirakawa, M.; Nakabeppu, Y. A molecular basis for the selective recognition of 2-hydroxy-dATP and 8-oxo-dGTP by human MTH1. J. Biol. Chem. 2002, 277, 8579–8587. [Google Scholar]
- Hayakawa, H.; Hofer, A.; Thelander, L.; Kitajima, S.; Cai, Y.; Oshiro, S.; Yakushiji, H.; Nakabeppu, Y.; Kuwano, M.; Sekiguchi, M. Metabolic fate of oxidized guanine ribonucleotides in mammalian cells. Biochemistry 1999, 38, 3610–3614. [Google Scholar] [CrossRef]
- Oda, H.; Taketomi, A.; Maruyama, R.; Itoh, R.; Nishioka, K.; Yakushiji, H.; Suzuki, T.; Sekiguchi, M.; Nakabeppu, Y. Multi-forms of human MTH1 polypeptides produced by alternative translation initiation and single nucleotide polymorphism. Nucleic Acids Res. 1999, 27, 4335–4343. [Google Scholar] [CrossRef]
- Kang, D.; Nishida, J.; Iyama, A.; Nakabeppu, Y.; Furuichi, M.; Fujiwara, T.; Sekiguchi, M.; Takeshige, K. Intracellular localization of 8-oxo-dGTPase in human cells, with special reference to the role of the enzyme in mitochondria. J. Biol. Chem. 1995, 270, 14659–14665. [Google Scholar]
- Radicella, J.P.; Dherin, C.; Desmaze, C.; Fox, M.S.; Boiteux, S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 8010–8015. [Google Scholar] [CrossRef]
- Lu, R.; Nash, H.; Verdine, G. A mammalian DNA repair enzyme that excises oxidatively damaged guanines maps to a locus frequently lost in lung cancer. Curr. Biol. 1977, 7, 397–407. [Google Scholar]
- Nishioka, K.; Ohtsubo, T.; Oda, H.; Fujiwara, T.; Kang, D.; Sugimachi, K.; Nakabeppu, Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell 1999, 10, 1637–1652. [Google Scholar] [CrossRef]
- Slupska, M.M.; Luther, W.M.; Chiang, J.H.; Yang, H.; Miller, J.H. Functional expression of hMYH, a human homolog of the Escherichia coli MutY protein. J. Bacteriol. 1999, 181, 6210–6213. [Google Scholar]
- Ohtsubo, T.; Nishioka, K.; Imaiso, Y.; Iwai, S.; Shimokawa, H.; Oda, H.; Fujiwara, T.; Nakabeppu, Y. Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res. 2000, 28, 1355–1364. [Google Scholar] [CrossRef]
- Parker, A.; Gu, Y.; Mahoney, W.; Lee, S.H.; Singh, K.K.; Lu, A.L. Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long patch DNA base excision repair. J. Biol. Chem. 2001, 276, 5547–5555. [Google Scholar]
- Hayashi, H.; Tominaga, Y.; Hirano, S.; McKenna, A.E.; Nakabeppu, Y.; Matsumoto, Y. Replication-associated repair of adenine:8-oxoguanine mispairs by MYH. Curr. Biol. 2002, 12, 335–339. [Google Scholar] [CrossRef]
- Hirano, S.; Tominaga, Y.; Ichinoe, A.; Ushijima, Y.; Tsuchimoto, D.; Honda-Ohnishi, Y.; Ohtsubo, T.; Sakumi, K.; Nakabeppu, Y. Mutator phenotype of MUTYH-null mouse embryonic stem cells. J. Biol. Chem. 2003, 278, 38121–38124. [Google Scholar] [CrossRef]
- Gu, Y.; Parker, A.; Wilson, T.M.; Bai, H.; Chang, D.Y.; Lu, A.L. Human MutY homolog, a DNA glycosylase involved in base excision repair, physically and functionally interacts with mismatch repair proteins human MutS homolog 2/human MutS homolog 6. J. Biol. Chem. 2002, 277, 11135–11142. [Google Scholar]
- Slupska, M.M.; Baikalov, C.; Luther, W.M.; Chiang, J.H.; Wei, Y.F.; Miller, J.H. Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J. Bacteriol. 1996, 178, 3885–3892. [Google Scholar]
- Tsuzuki, T.; Egashira, A.; Igarashi, H.; Iwakuma, T.; Nakatsuru, Y.; Tominaga, Y.; Kawate, H.; Nakao, K.; Nakamura, K.; Ide, F.; et al. Spontaneous tumorigenesis in mice defective in the MTH1 gene encoding 8-oxo-dGTPase. Proc. Natl. Acad. Sci. USA 2001, 98, 11456–11461. [Google Scholar] [CrossRef]
- Sakumi, K.; Tominaga, Y.; Furuichi, M.; Xu, P.; Tsuzuki, T.; Sekiguchi, M.; Nakabeppu, Y. Ogg1 knockout-associated lung tumorigenesis and its suppression by Mth1 gene disruption. Cancer Res. 2003, 63, 902–905. [Google Scholar]
- Klungland, A.; Rosewell, I.; Hollenbach, S.; Larsen, E.; Daly, G.; Epe, B.; Seeberg, E.; Lindahl, T.; Barnes, D.E. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl. Acad. Sci. USA 1999, 96, 13300–13305. [Google Scholar] [CrossRef]
- Minowa, O.; Arai, T.; Hirano, M.; Monden, Y.; Nakai, S.; Fukuda, M.; Itoh, M.; Takano, H.; Hippou, Y.; Aburatani, H.; et al. Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 4156–4161. [Google Scholar] [CrossRef]
- Chevillard, S.; Radicella, J.; Levalois, C.; Lebeau, J.; Poupon, M.; Oudard, S.; Dutrillaux, B.; Boiteux, S. Mutations in OGG1, a gene involved in the repair of oxidative DNA damage, are found in human lung and kidney tumours. Oncogene 1998, 16, 3083–3086. [Google Scholar]
- Sakamoto, K.; Tominaga, Y.; Yamauchi, K.; Nakatsu, Y.; Sakumi, K.; Yoshiyama, K.; Egashira, A.; Kura, S.; Yao, T.; Tsuneyoshi, M.; et al. MUTYH-null mice are susceptible to spontaneous and oxidative stress induced intestinal tumorigenesis. Cancer Res. 2007, 67, 6599–6604. [Google Scholar] [CrossRef]
- Al-Tassan, N.; Chmiel, N.H.; Maynard, J.; Fleming, N.; Livingston, A.L.; Williams, G.T.; Hodges, A.K.; Davies, D.R.; David, S.S.; Sampson, J.R.; et al. Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat. Genet. 2002, 30, 227–232. [Google Scholar] [CrossRef]
- Nielsen, M.; Morreau, H.; Vasen, H.F.; Hes, F.J. MUTYH-associated polyposis (MAP). Crit. Rev. Oncol. Hematol. 2011, 79, 1–16. [Google Scholar] [CrossRef]
- Xie, Y.; Yang, H.; Cunanan, C.; Okamoto, K.; Shibata, D.; Pan, J.; Barnes, D.E.; Lindahl, T.; McIlhatton, M.; Fishel, R.; et al. Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K-ras oncogene in lung tumors. Cancer Res. 2004, 64, 3096–3102. [Google Scholar] [CrossRef]
- Ohno, M.; Sakumi, K.; Fukumura, R.; Furuichi, M.; Iwasaki, Y.; Hokama, M.; Ikemura, T.; Tsuzuki, T.; Gondo, Y.; Nakabeppu, Y. 8-Oxoguanine causes spontaneous de novo germline mutations in mice. Sci. Rep. 2014, 4, 4689. [Google Scholar]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar]
- Kong, A.; Frigge, M.L.; Masson, G.; Besenbacher, S.; Sulem, P.; Magnusson, G.; Gudjonsson, S.A.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 2012, 488, 471–475. [Google Scholar]
- Tajiri, T.; Maki, H.; Sekiguchi, M. Functional cooperation of MutT, MutM and MutY proteins in preventing mutations caused by spontaneous oxidation of guanine nucleotide in Escherichia coli. Mutat. Res. 1995, 336, 257–267. [Google Scholar] [CrossRef]
- Russo, M.T.; Blasi, M.F.; Chiera, F.; Fortini, P.; Degan, P.; Macpherson, P.; Furuichi, M.; Nakabeppu, Y.; Karran, P.; Aquilina, G.; et al. The oxidized deoxynucleoside triphosphate pool is a significant contributor to genetic instability in mismatch repair-deficient cells. Mol. Cell. Biol. 2004, 24, 465–474. [Google Scholar] [CrossRef]
- Arai, T.; Kelly, V.P.; Minowa, O.; Noda, T.; Nishimura, S. High accumulation of oxidative DNA damage, 8-hydroxyguanine, in Mmh/Ogg1 deficient mice by chronic oxidative stress. Carcinogenesis 2002, 23, 2005–2010. [Google Scholar] [CrossRef]
- Arai, T.; Kelly, V.P.; Minowa, O.; Noda, T.; Nishimura, S. The study using wild-type and Ogg1 knockout mice exposed to potassium bromate shows no tumor induction despite an extensive accumulation of 8-hydroxyguanine in kidney DNA. Toxicology 2006, 221, 179–186. [Google Scholar] [CrossRef]
- Doucas, H.; Garcea, G.; Neal, C.P.; Manson, M.M.; Berry, D.P. Changes in the Wnt signalling pathway in gastrointestinal cancers and their prognostic significance. Eur. J. Cancer 2005, 41, 365–379. [Google Scholar] [CrossRef]
- Tsuzuki, T.; Piao, J.S.; Isoda, T.; Sakumi, K.; Nakabeppu, Y.; Nakatsu, Y. Oxidative stress-induced tumorigenesis in the small intestines of DNA repair-deficient mice. Health Phys. 2011, 100, 293–294. [Google Scholar] [CrossRef]
- Takahashi-Yanaga, F.; Yoshihara, T.; Jingushi, K.; Igawa, K.; Tomooka, K.; Watanabe, Y.; Morimoto, S.; Nakatsu, Y.; Tsuzuki, T.; Nakabeppu, Y.; et al. DIF-1 inhibits tumor growth in vivo reducing phosphorylation of GSK-3β and expressions of cyclin D1 and TCF7L2 in cancer model mice. Biochem. Pharmacol. 2014, 89, 340–348. [Google Scholar] [CrossRef]
- Kunisada, M.; Sakumi, K.; Tominaga, Y.; Budiyanto, A.; Ueda, M.; Ichihashi, M.; Nakabeppu, Y.; Nishigori, C. 8-Oxoguanine formation induced by chronic UVB exposure makes Ogg1 knockout mice susceptible to skin carcinogenesis. Cancer Res. 2005, 65, 6006–6010. [Google Scholar] [CrossRef]
- Yogianti, F.; Kunisada, M.; Ono, R.; Sakumi, K.; Nakabeppu, Y.; Nishigori, C. Skin tumours induced by narrowband UVB have higher frequency of p53 mutations than tumours induced by broadband UVB independent of Ogg1 genotype. Mutagenesis 2012, 27, 637–643. [Google Scholar] [CrossRef]
- Yoshimura, D.; Sakumi, K.; Ohno, M.; Sakai, Y.; Furuichi, M.; Iwai, S.; Nakabeppu, Y. An oxidized purine nucleoside triphosphatase, MTH1, suppresses cell death caused by oxidative stress. J. Biol. Chem. 2003, 278, 37965–37973. [Google Scholar]
- Ichikawa, J.; Tsuchimoto, D.; Oka, S.; Ohno, M.; Furuichi, M.; Sakumi, K.; Nakabeppu, Y. Oxidation of mitochondrial deoxynucleotide pools by exposure to sodium nitroprusside induces cell death. DNA Repair 2008, 7, 418–430. [Google Scholar]
- De Luca, G.; Russo, M.T.; Degan, P.; Tiveron, C.; Zijno, A.; Meccia, E.; Ventura, I.; Mattei, E.; Nakabeppu, Y.; Crescenzi, M.; et al. A role for oxidized DNA precursors in Huntington’s disease-like striatal neurodegeneration. PLoS Genet. 2008, 4, e1000266. [Google Scholar]
- Oka, S.; Ohno, M.; Tsuchimoto, D.; Sakumi, K.; Furuichi, M.; Nakabeppu, Y. Two distinct pathways of cell death triggered by oxidative damage to nuclear and mitochondrial DNAs. EMBO J. 2008, 27, 421–432. [Google Scholar] [CrossRef]
- Hashimoto, K.; Tominaga, Y.; Nakabeppu, Y.; Moriya, M. Futile short-patch DNA base excision repair of adenine: 8-Oxoguanine mispair. Nucleic Acids Res. 2004, 32, 5928–5934. [Google Scholar] [CrossRef]
- Gad, H.; Koolmeister, T.; Jemth, A.S.; Eshtad, S.; Jacques, S.A.; Strom, C.E.; Svensson, L.M.; Schultz, N.; Lundback, T.; Einarsdottir, B.O.; et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014, 508, 215–221. [Google Scholar] [CrossRef]
- Huber, K.V.; Salah, E.; Radic, B.; Gridling, M.; Elkins, J.M.; Stukalov, A.; Jemth, A.S.; Gokturk, C.; Sanjiv, K.; Stromberg, K.; et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature 2014, 508, 222–227. [Google Scholar] [CrossRef]
- Toyokuni, S.; Okamoto, K.; Yodoi, J.; Hiai, H. Persistent oxidative stress in cancer. FEBS Lett. 1995, 358, 1–3. [Google Scholar]
- Kennedy, C.H.; Cueto, R.; Belinsky, S.A.; Lechner, J.F.; Pryor, W.A. Overexpression of hMTH1 mRNA: A molecular marker of oxidative stress in lung cancer cells. FEBS Lett. 1998, 429, 17–20. [Google Scholar] [CrossRef]
- Iida, T.; Furuta, A.; Kawashima, M.; Nishida, J.; Nakabeppu, Y.; Iwaki, T. Accumulation of 8-oxo-2'-deoxyguanosine and increased expression of hMTH1 protein in brain tumors. Neuro-Oncology 2001, 3, 73–81. [Google Scholar]
- Kennedy, C.H.; Pass, H.I.; Mitchell, J.B. Expression of human MutT homologue (hMTH1) protein in primary non-small-cell lung carcinomas and histologically normal surrounding tissue. Free Radic. Biol. Med. 2003, 34, 1447–1457. [Google Scholar] [CrossRef]
- Oncomine. Available online: https://www.oncomine.org (accessed on 14 July 2014).
- Rai, P.; Onder, T.T.; Young, J.J.; McFaline, J.L.; Pang, B.; Dedon, P.C.; Weinberg, R.A. Continuous elimination of oxidized nucleotides is necessary to prevent rapid onset of cellular senescence. Proc. Natl. Acad. Sci. USA 2009, 106, 169–174. [Google Scholar]
- Sheng, Z.; Oka, S.; Tsuchimoto, D.; Abolhassani, N.; Nomaru, H.; Sakumi, K.; Yamada, H.; Nakabeppu, Y. 8-Oxoguanine causes neurodegeneration during MUTYH-mediated DNA base excision repair. J. Clin. Investig. 2012, 122, 4344–4361. [Google Scholar] [CrossRef]
- Kajitani, K.; Yamaguchi, H.; Dan, Y.; Furuichi, M.; Kang, D.; Nakabeppu, Y. MTH1, an oxidized purine nucleoside triphosphatase, suppresses the accumulation of oxidative damage of nucleic acids in the hippocampal microglia during kainate-induced excitotoxicity. J. Neurosci. 2006, 26, 1688–1698. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Kajitani, K.; Dan, Y.; Furuichi, M.; Ohno, M.; Sakumi, K.; Kang, D.; Nakabeppu, Y. MTH1, an oxidized purine nucleoside triphosphatase, protects the dopamine neurons from oxidative damage in nucleic acids caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Cell Death Differ. 2006, 13, 551–563. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nakabeppu, Y. Cellular Levels of 8-Oxoguanine in either DNA or the Nucleotide Pool Play Pivotal Roles in Carcinogenesis and Survival of Cancer Cells. Int. J. Mol. Sci. 2014, 15, 12543-12557. https://doi.org/10.3390/ijms150712543
Nakabeppu Y. Cellular Levels of 8-Oxoguanine in either DNA or the Nucleotide Pool Play Pivotal Roles in Carcinogenesis and Survival of Cancer Cells. International Journal of Molecular Sciences. 2014; 15(7):12543-12557. https://doi.org/10.3390/ijms150712543
Chicago/Turabian StyleNakabeppu, Yusaku. 2014. "Cellular Levels of 8-Oxoguanine in either DNA or the Nucleotide Pool Play Pivotal Roles in Carcinogenesis and Survival of Cancer Cells" International Journal of Molecular Sciences 15, no. 7: 12543-12557. https://doi.org/10.3390/ijms150712543
APA StyleNakabeppu, Y. (2014). Cellular Levels of 8-Oxoguanine in either DNA or the Nucleotide Pool Play Pivotal Roles in Carcinogenesis and Survival of Cancer Cells. International Journal of Molecular Sciences, 15(7), 12543-12557. https://doi.org/10.3390/ijms150712543