The Critical Role of Membrane Cholesterol in Salmonella-Induced Autophagy in Intestinal Epithelial Cells


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
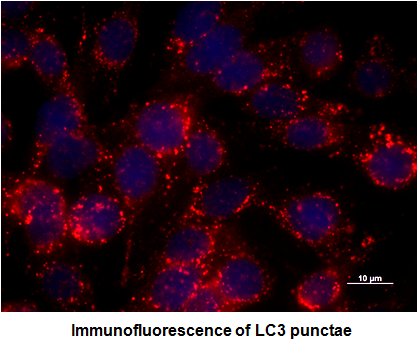
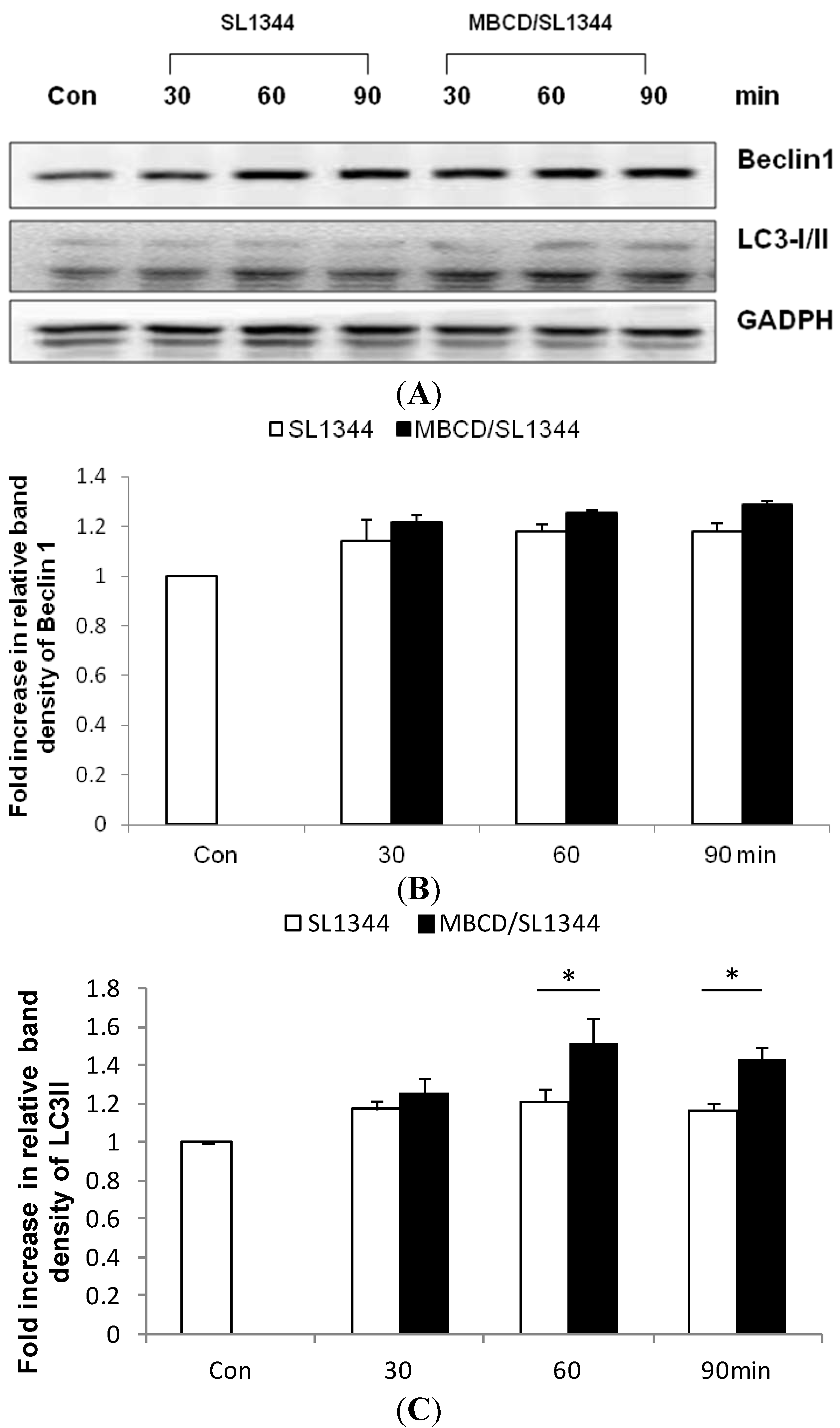
2.1. Depletion of Membrane Cholesterol Induced the Activation of Autophagy Proteins Expression in Salmonella-Infected Intestinal Epithelial Cell (IEC)

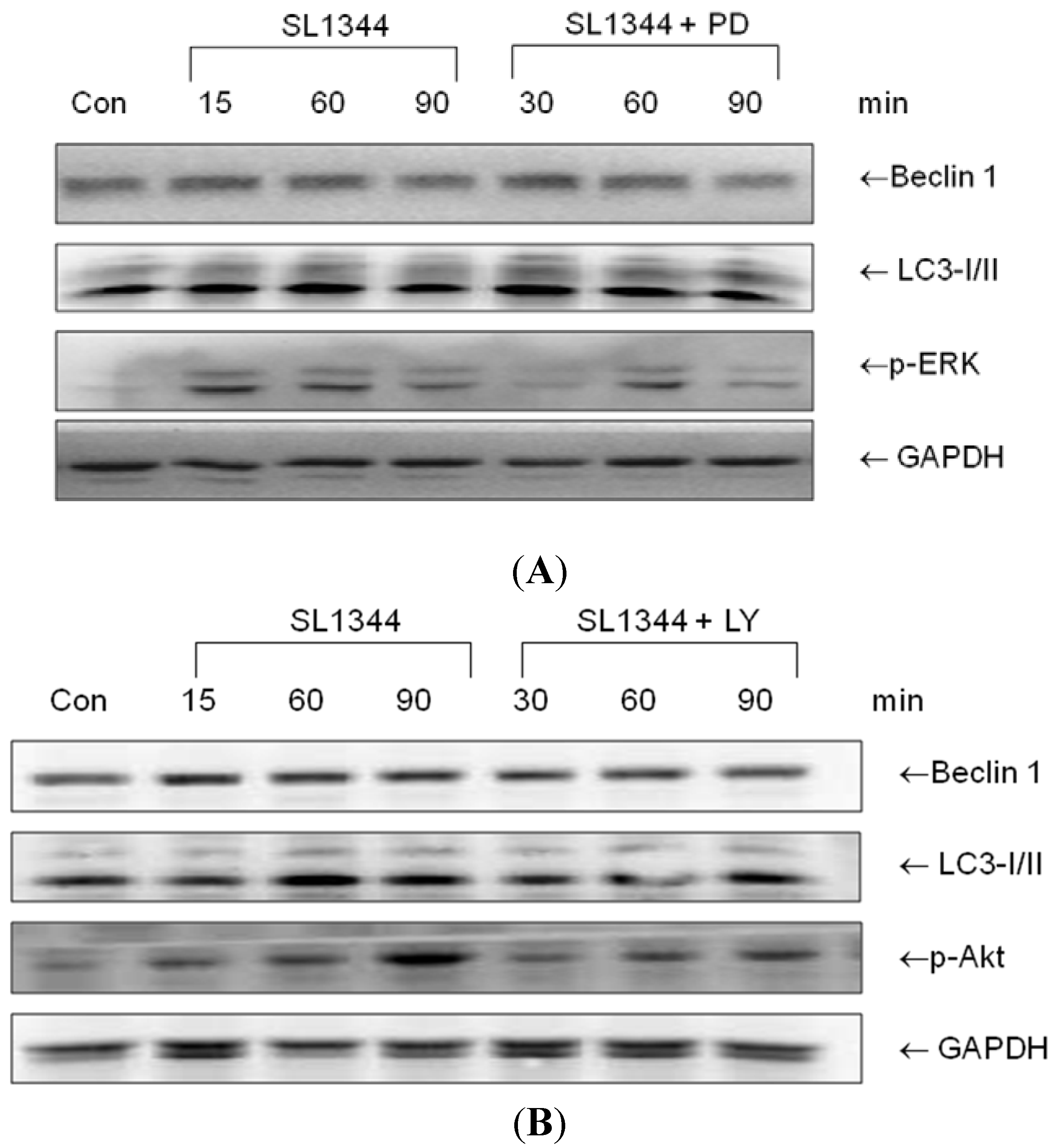
2.2. The Involvement of ERK and Akt in Beclin 1 or LC3II Proteins Expression in Salmonella-Infected IEC

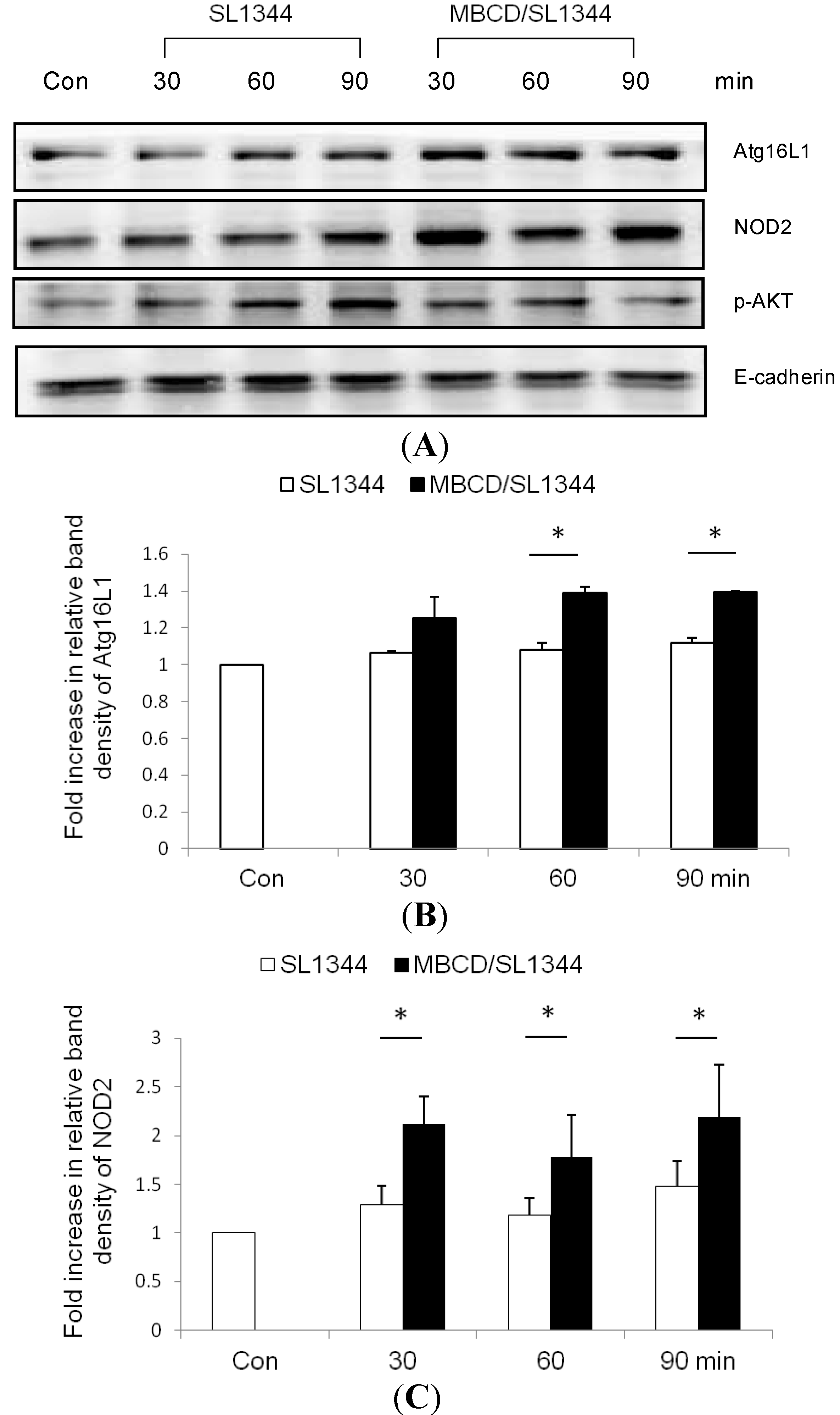
2.3. The Membrane Association of Nucleotide-Binding Oligomerization Domain-Containing Protein 2 (NOD2) and Atg16L1 Is Affected by the Depletion of Membrane Cholesterol

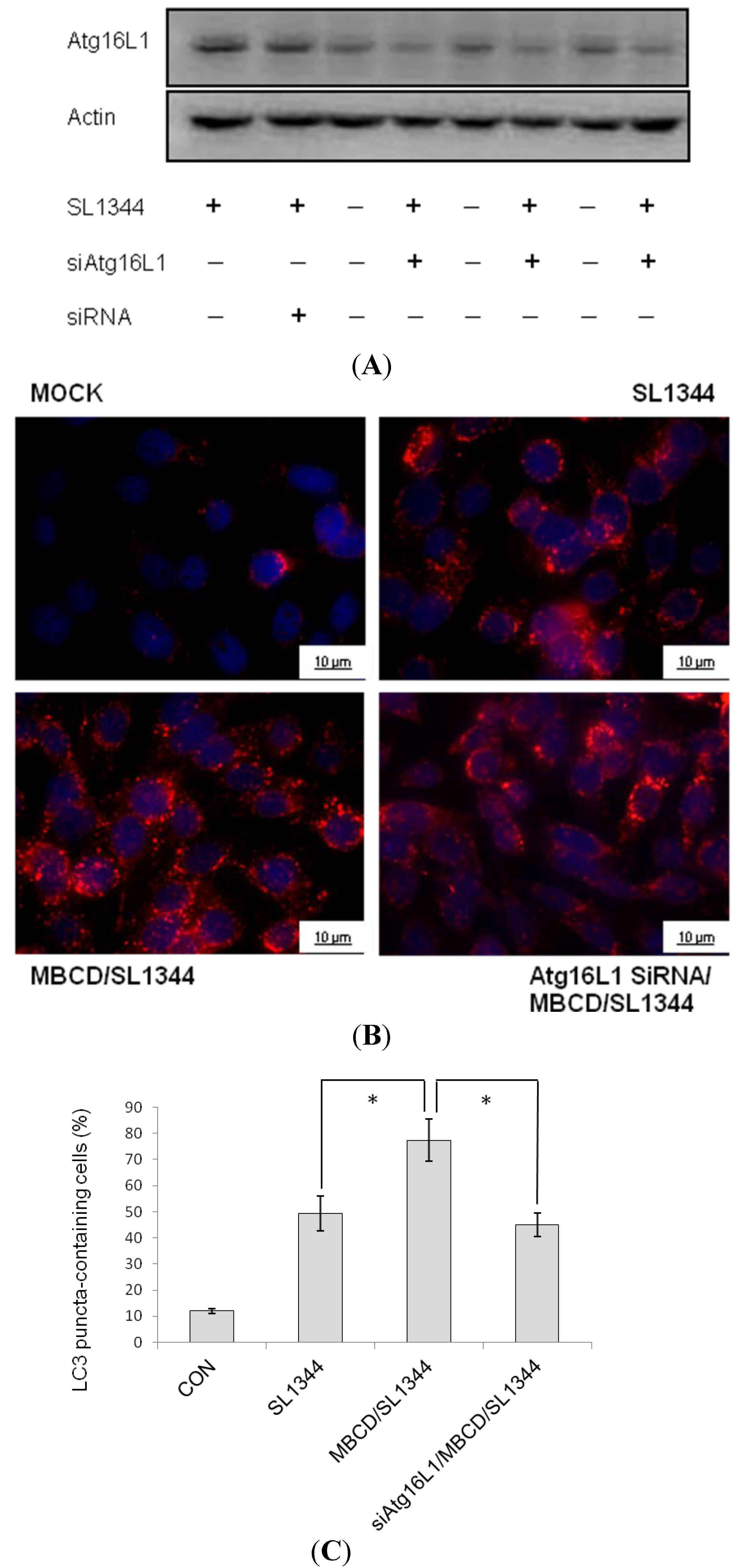
2.4. Involvement of Atg16L1 in Enhancement of Salmonella-Induced Autophagy by Methyl-beta-cyclodextrin (MBCD)
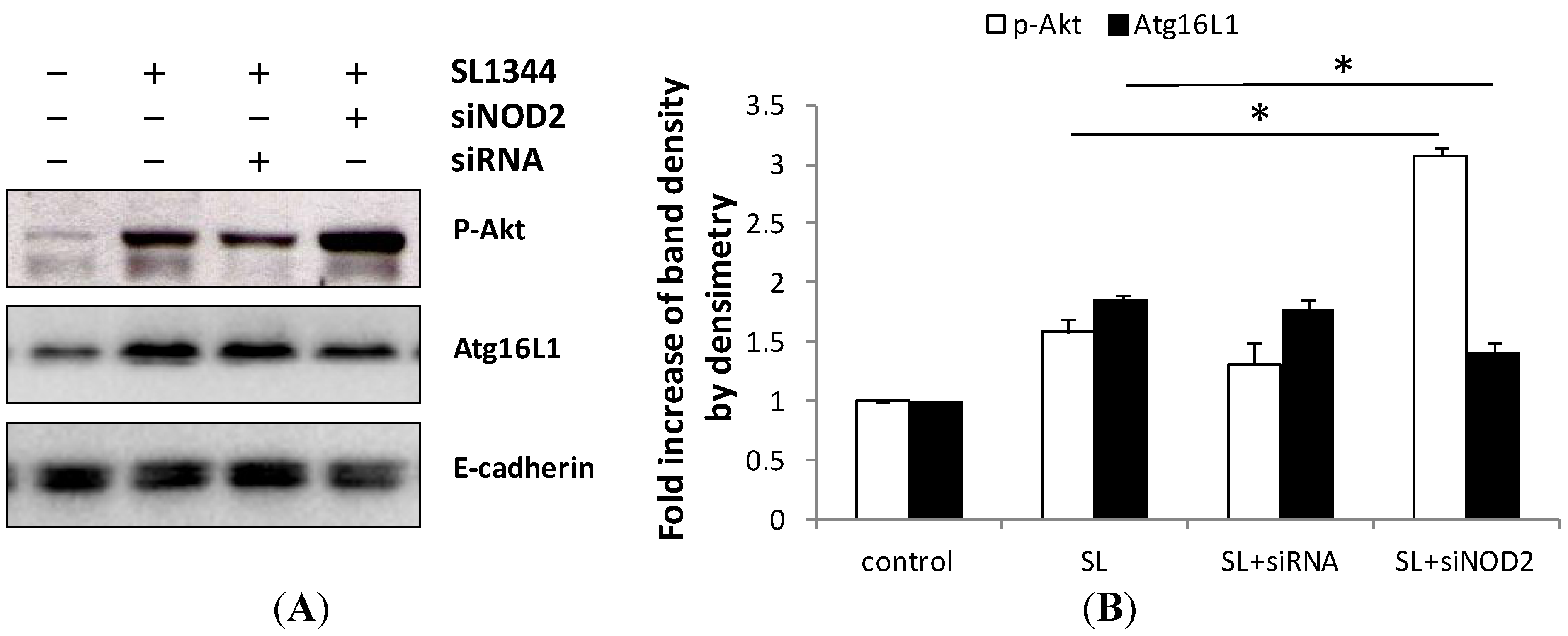
2.5. Activation of NOD2 Suppressed the Activation of Akt in Salmonella-Infected Caco-2 Cells


2.6. Discussion
3. Experimental Section
3.1. Reagents
3.2. Bacterial Strains
3.3. Cell Culture and Infection
3.4. Depletion of Cellular Cholesterol
3.5. Cell Fractionation
3.6. Western Blot
3.7. Real-Time PCR for mRNA Assay
3.8. Small-Interfering RNA (siRNA) Transfection
3.9. Immunofluorescence Analysis
3.10. Cell Viability and Morphologic Features
3.11. Statistical Analysis
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Conflicts of Interest
References
- Deretic, V. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol. Rev. 2011, 240, 92–104. [Google Scholar]
- Moreau, K.; Rubinsztein, D.C. The plasma membrane as a control center for autophagy. Autophagy 2012, 8, 861–863. [Google Scholar]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar]
- Knaevelsrud, H.; Simonsen, A. Lipids in autophagy: Constituents, signaling molecules and cargo with relevance to disease. Biochim. Biophys. Acta 2012, 1821, 1133–1145. [Google Scholar]
- Huang, F.C. Plasma membrane cholesterol plays a critical role in the Salmonella-induced anti-inflammatory response in intestinal epithelial cells. Cell. Immunol. 2011, 271, 480–487. [Google Scholar]
- Huang, F.C.; Li, Q.; Cherayil, B.J. A phosphatidyl-inositol-3-kinase-dependent anti-inflammatory pathway activated by Salmonella in epithelial cells. FEMS Microbiol. Lett. 2005, 243, 265–270. [Google Scholar]
- Birmingham, C.L.; Brumell, J.H. Autophagy recognizes intracellular Salmonella enterica serovar Typhimurium in damaged vacuoles. Autophagy 2006, 2, 156–158. [Google Scholar]
- Noda, T.; Kageyama, S.; Fujita, N.; Yoshimori, T. Three-axis model for Atg recruitment in autophagy against Salmonella. Int. J. Cell Biol. 2012, 2012, 389562. [Google Scholar]
- Ellington, A.A.; Berhow, M.A.; Singletary, K.W. Inhibition of Akt signaling and enhanced ERK1/2 activity are involved in induction of macroautophagy by triterpenoid B-group soyasaponins in colon cancer cells. Carcinogenesis 2006, 27, 298–306. [Google Scholar]
- Ogier-Denis, E.; Pattingre, S.; El Benna, J.; Codogno, P. Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J. Biol. Chem. 2000, 275, 39090–39095. [Google Scholar]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.G.; Magalhaes, J.G.; Yuan, L.; Soares, F.; Chea, E.; Le Bourhis, L.; et al. NOD1 and NOD2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar]
- Cooney, R.; Baker, J.; Brain, O.; Danis, B.; Pichulik, T.; Allan, P.; Ferguson, D.J.; Campbell, B.J.; Jewell, D.; Simmons, A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 2010, 16, 90–97. [Google Scholar]
- Lapaquette, P.; Darfeuille-Michaud, A. Abnormalities in the handling of intracellular bacteria in Crohn’s disease. J. Clin. Gastroenterol. 2010, 44, S26–S29. [Google Scholar]
- Kuballa, P.; Huett, A.; Rioux, J.D.; Daly, M.J.; Xavier, R.J. Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS One 2008, 3, e3391. [Google Scholar]
- Homer, C.R.; Richmond, A.L.; Rebert, N.A.; Achkar, J.P.; McDonald, C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology 2010, 139, 1630–1641. [Google Scholar]
- Messer, J.S.; Murphy, S.F.; Logsdon, M.F.; Lodolce, J.P.; Grimm, W.A.; Bartulis, S.J.; Vogel, T.P.; Burn, M.; Boone, D.L. The Crohn’s disease: Associated ATG16L1 variant and Salmonella invasion. BMJ Open 2014. [Google Scholar] [CrossRef]
- Billmann-Born, S.; Lipinski, S.; Bock, J.; Till, A.; Rosenstiel, P.; Schreiber, S. The complex interplay of NOD-like receptors and the autophagy machinery in the pathophysiology of Crohn disease. Eur. J. Cell Biol. 2011, 90, 593–602. [Google Scholar]
- Arico, S.; Petiot, A.; Bauvy, C.; Dubbelhuis, P.F.; Meijer, A.J.; Codogno, P.; Ogier-Denis, E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2001, 276, 35243–35246. [Google Scholar]
- Scarlatti, F.; Bauvy, C.; Ventruti, A.; Sala, G.; Cluzeaud, F.; Vandewalle, A.; Ghidoni, R.; Codogno, P. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of Beclin 1. J. Biol. Chem. 2004, 279, 18384–18391. [Google Scholar]
- Wang, J.; Whiteman, M.W.; Lian, H.; Wang, G.; Singh, A.; Huang, D.; Denmark, T. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J. Biol. Chem. 2009, 284, 21412–21424. [Google Scholar]
- Liu, Y.; Yang, Y.; Ye, Y.C.; Shi, Q.F.; Chai, K.; Tashiro, S.; Onodera, S.; Ikejima, T. Activation of ERK-p53 and ERK-mediated phosphorylation of Bcl-2 are involved in autophagic cell death induced by the c-Met inhibitor SU11274 in human lung cancer A549 cells. J. Pharmacol. Sci. 2012, 118, 423–432. [Google Scholar]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar]
- Huang, F.C. Regulation of Salmonella flagellin-induced interleukin-8 in intestinal epithelial cells by muramyl dipeptide. Cell Immunol. 2012, 278, 1–9. [Google Scholar]
- Cheng, J.; Ohsaki, Y.; Tauchi-Sato, K.; Fujita, A.; Fujimoto, T. Cholesterol depletion induces autophagy. Biochem. Biophys. Res. Commun. 2006, 351, 246–252. [Google Scholar]
- Benjamin, J.L.; Sumpter, R., Jr.; Levine, B.; Hooper, L.V. Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe 2013, 13, 723–734. [Google Scholar]
- Shi, C.S.; Kehrl, J.H. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 2008, 283, 33175–33182. [Google Scholar]
- Codogno, P.; Mehrpour, M.; Proikas-Cezanne, T. Canonical and non-canonical autophagy: Variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol. 2012, 13, 7–12. [Google Scholar]
- Gelman, A.E.; LaRosa, D.F.; Zhang, J.; Walsh, P.T.; Choi, Y.; Sunyer, J.O.; Turka, L.A. The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity 2006, 25, 783–793. [Google Scholar]
- Smith, K.A.; Maizels, R.M. Defeating sepsis by misleading MyD88. Nat. Immunol. 2011, 12, 284–286. [Google Scholar]
- Conway, K.L.; Kuballa, P.; Song, J.H.; Patel, K.K.; Castoreno, A.B.; Yilmaz, O.H.; Jijon, H.B.; Zhang, M.; Aldrich, L.N.; Villablanca, E.J.; et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology 2013, 145, 1347–1357. [Google Scholar]
- De Chastellier, C.; Thilo, L. Cholesterol depletion in Mycobacterium avium-infected macrophages overcomes the block in phagosome maturation and leads to the reversible sequestration of viable mycobacteria in phagolysosome-derived autophagic vacuoles. Cell. Microbiol. 2006, 8, 242–256. [Google Scholar]
- Kumar, Y.; Valdivia, R.H. Leading a sheltered life: Intracellular pathogens and maintenance of vacuolar compartments. Cell Host Microbe 2009, 5, 593–601. [Google Scholar]
- Birmingham, C.L.; Smith, A.C.; Bakowski, M.A.; Yoshimori, T.; Brumell, J.H. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem. 2006, 281, 11374–11383. [Google Scholar]
- Jia, K.; Thomas, C.; Akbar, M.; Sun, Q.; Adams-Huet, B.; Gilpin, C.; Levine, B. Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling-regulated pathogen resistance. Proc. Natl. Acad. Sci. USA 2009, 106, 14564–14569. [Google Scholar]
- Garcia Rodriguez, L.A.; Ruigomez, A.; Panes, J. Acute gastroenteritis is followed by an increased risk of inflammatory bowel disease. Gastroenterology 2006, 130, 1588–1594. [Google Scholar]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar]
- Knodler, L.A.; Finlay, B.B.; Steele-Mortimer, O. The Salmonella effector protein SopB protects epithelial cells from apoptosis by sustained activation of Akt. J. Biol. Chem. 2005, 280, 9058–9064. [Google Scholar]
- Knodler, L.A.; Winfree, S.; Drecktrah, D.; Ireland, R.; Steele-Mortimer, O. Ubiquitination of the bacterial inositol phosphatase, SopB, regulates its biological activity at the plasma membrane. Cell. Microbiol. 2009, 11, 1652–1670. [Google Scholar]
- Huang, F.C. Upregulation of Salmonella-induced IL-6 production in Caco-2 cells by PJ-34, PARP-1 inhibitor: Involvement of PI3K, p38 MAPK, ERK, JNK, and NF-κB. Mediators Inflamm. 2009, 2009, 103890. [Google Scholar]
- Huang, F.C.; Werne, A.; Li, Q.; Galyov, E.E.; Walker, W.A.; Cherayil, B.J. Cooperative interactions between flagellin and SopE2 in the epithelial interleukin-8 response to Salmonella enterica serovar typhimurium infection. Infect. Immun. 2004, 72, 5052–5062. [Google Scholar]
- Christian, A.E.; Haynes, M.P.; Phillips, M.C.; Rothblat, G.H. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 1997, 38, 2264–2272. [Google Scholar]
- Scheid, M.P.; Marignani, P.A.; Woodgett, J.R. Multiple phosphoinositide 3-kinase-dependent steps in activation of protein kinase B. Mol. Cell. Biol. 2002, 22, 6247–6260. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, F.-C. The Critical Role of Membrane Cholesterol in Salmonella-Induced Autophagy in Intestinal Epithelial Cells. Int. J. Mol. Sci. 2014, 15, 12558-12572. https://doi.org/10.3390/ijms150712558
Huang F-C. The Critical Role of Membrane Cholesterol in Salmonella-Induced Autophagy in Intestinal Epithelial Cells. International Journal of Molecular Sciences. 2014; 15(7):12558-12572. https://doi.org/10.3390/ijms150712558
Chicago/Turabian StyleHuang, Fu-Chen. 2014. "The Critical Role of Membrane Cholesterol in Salmonella-Induced Autophagy in Intestinal Epithelial Cells" International Journal of Molecular Sciences 15, no. 7: 12558-12572. https://doi.org/10.3390/ijms150712558
APA StyleHuang, F. -C. (2014). The Critical Role of Membrane Cholesterol in Salmonella-Induced Autophagy in Intestinal Epithelial Cells. International Journal of Molecular Sciences, 15(7), 12558-12572. https://doi.org/10.3390/ijms150712558