Synthesis and Properties of Poly(l-lactide)-b-poly (l-phenylalanine) Hybrid Copolymers


Abstract
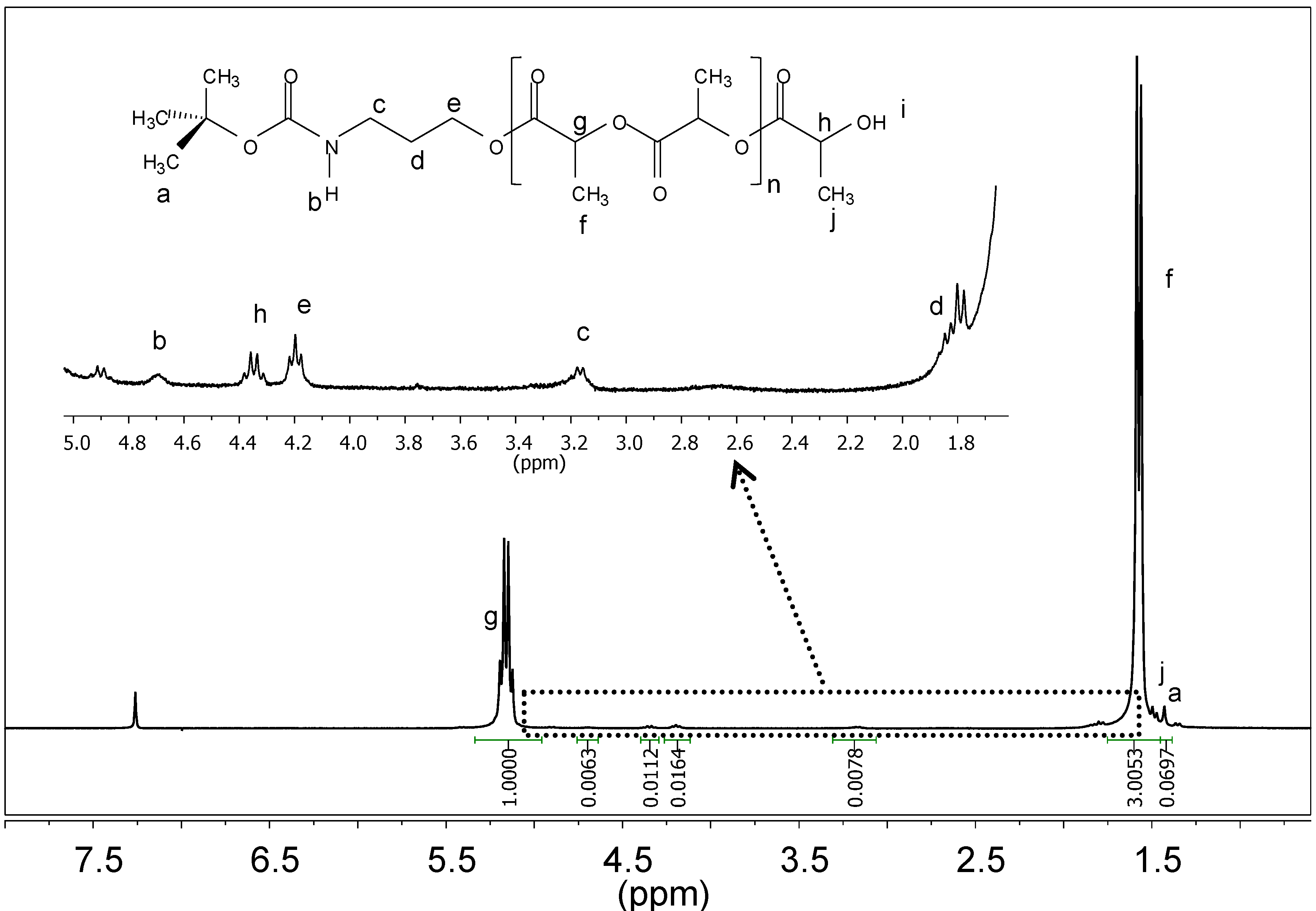
:
1. Introduction
2. Results and Discussion
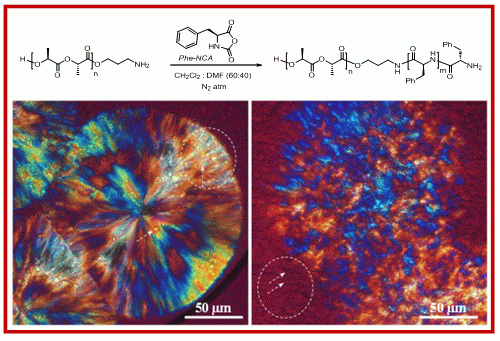
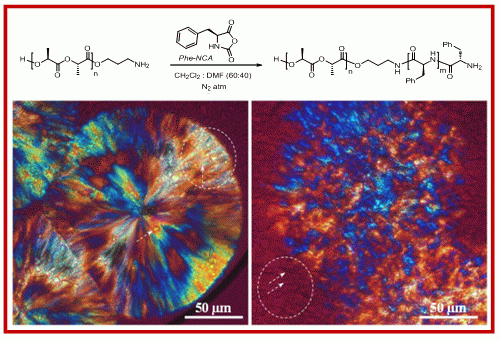
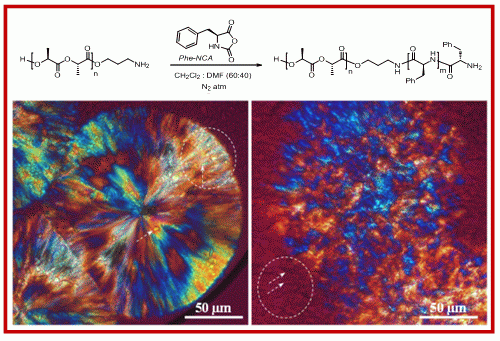
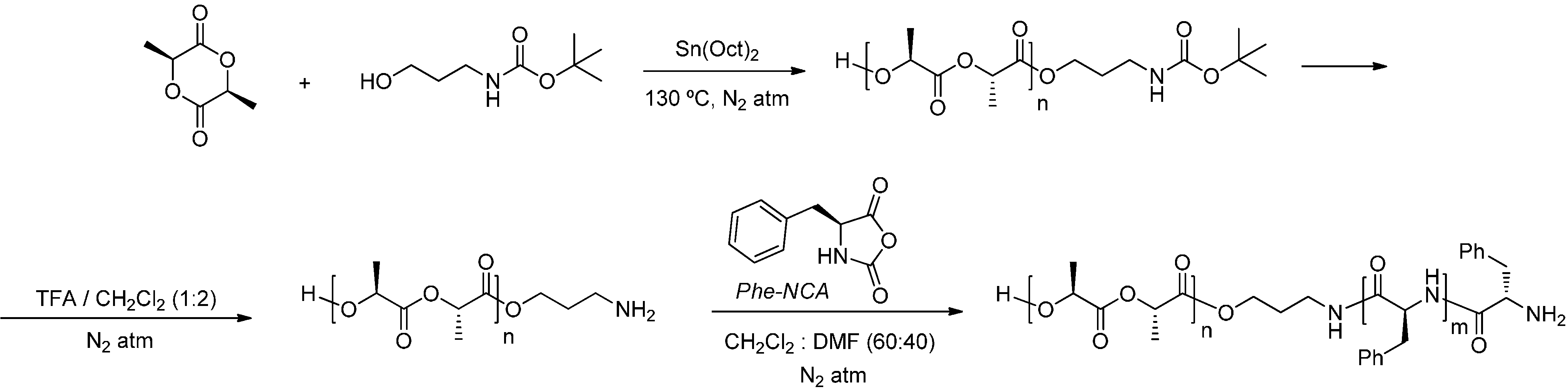
2.1. Synthesis of Hybrid Copolymers


{kind=link}
{kind=link}
{kind=link}
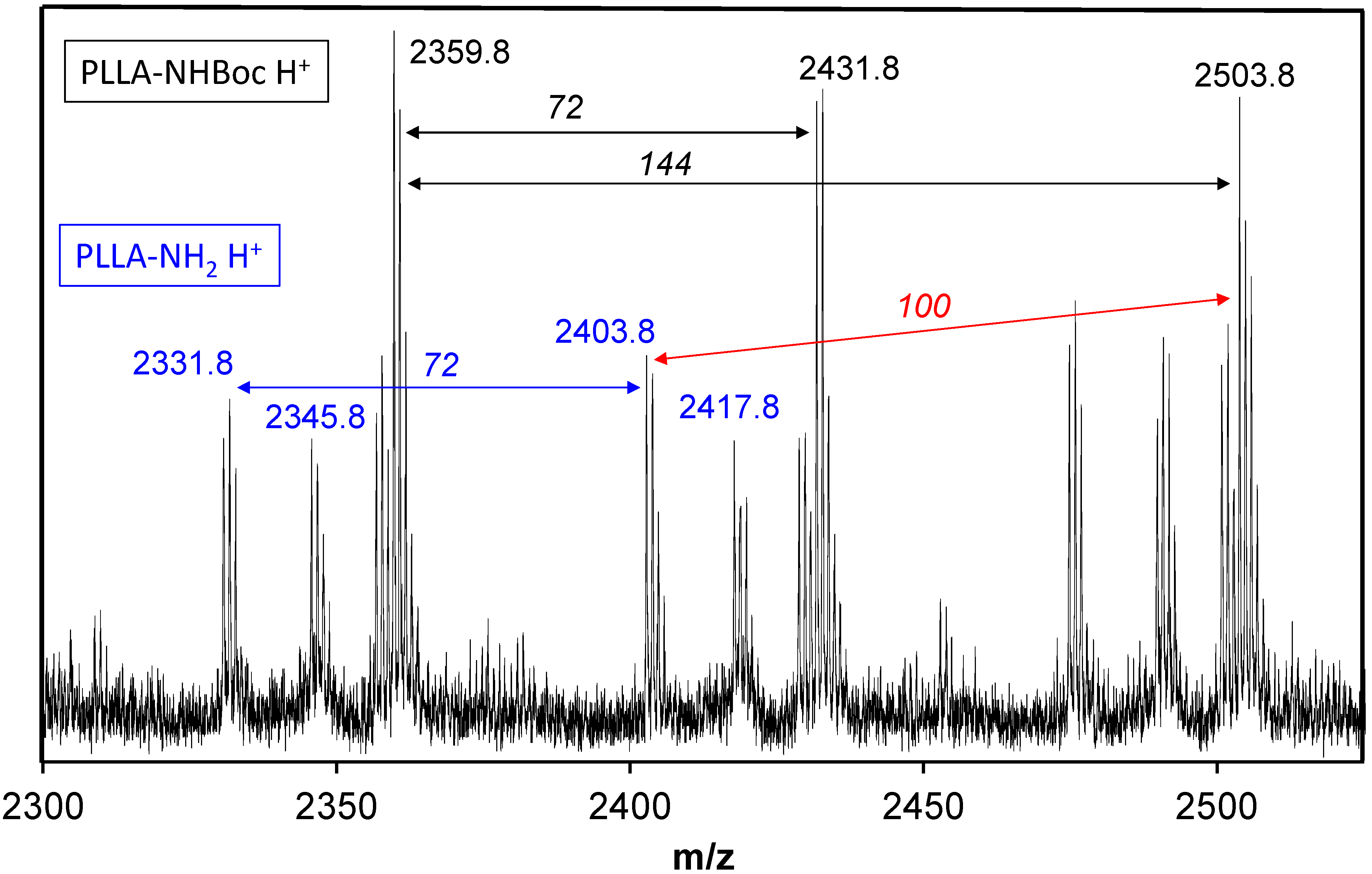{kind=link}
{kind=link}
{kind=link}
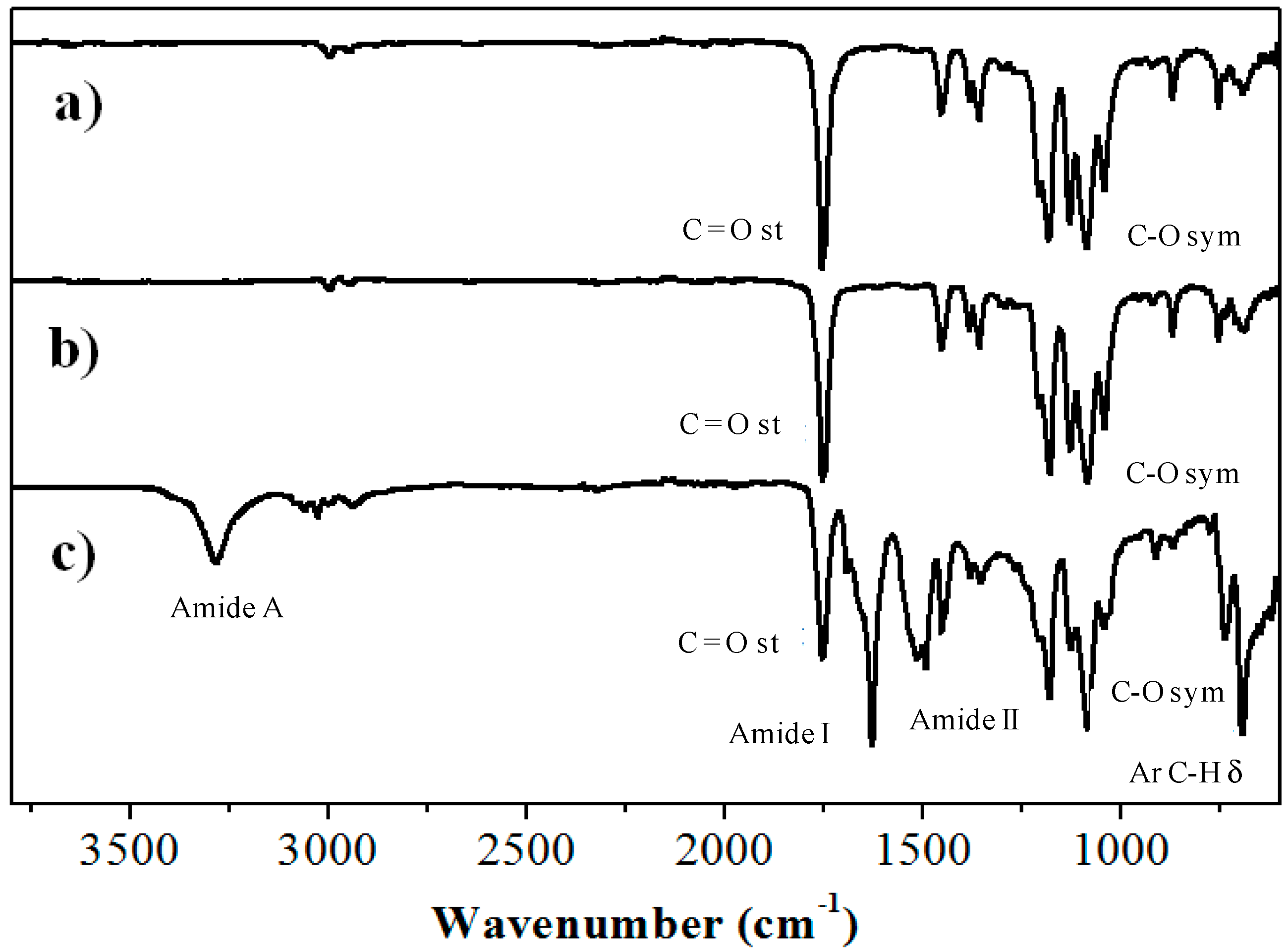{kind=link}
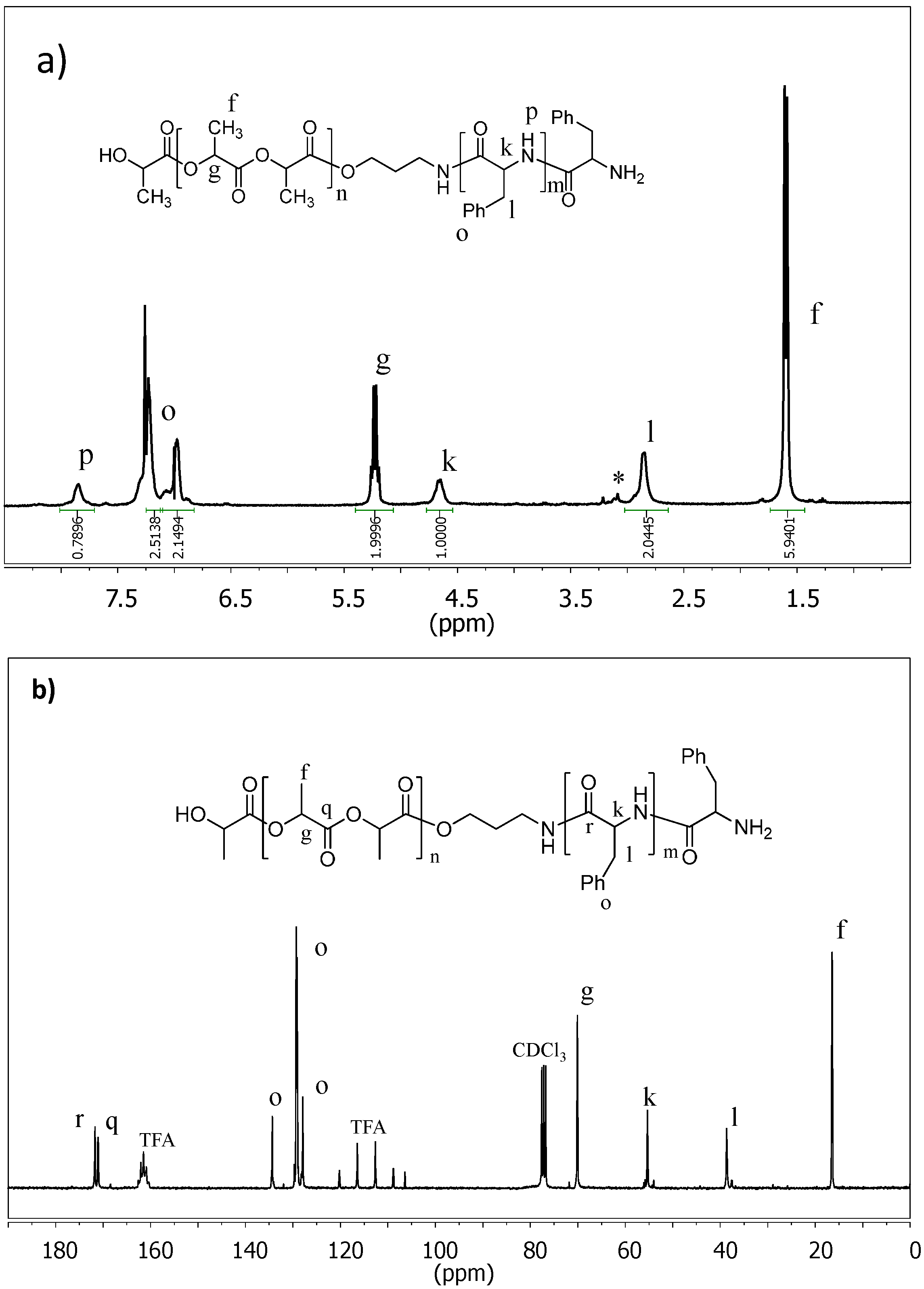{kind=link}
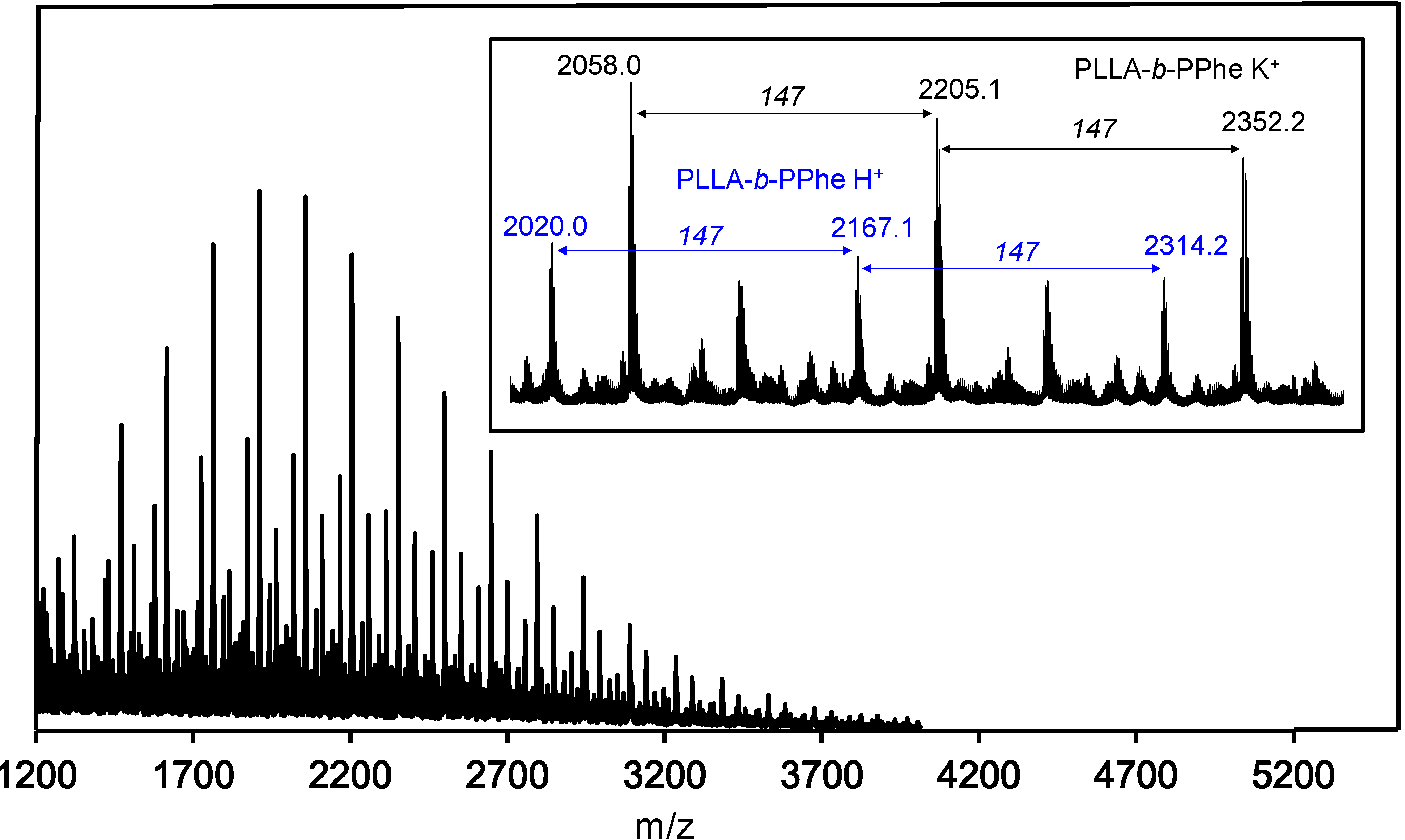{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mn a | Mn b | MW b | PDI b | Mn c | f d |
|---|---|---|---|---|---|---|
| PLLA10 | 1440 | 1120 | 3200 | 2.62 | 1285 | 87 |
| PLLA25 | 3600 | 3562 | 6105 | 1.71 | 3107 | 86 |
| PLLA50 | 7200 | 6978 | 14,958 | 2.14 | 3406 | 82 |
| PLLA100 | 14,400 | 13,235 | 17,217 | 1.30 | 8858 | 73 |



| Sample a | LLA:l-Phe b | LLA:l-Phe c | Mn of the PPhe Block | |
|---|---|---|---|---|
| Mn d | Mn c | |||
| PLLA50-b-PPhe10 | 83:17 | 83:17 | 1470 | 1506 |
| PLLA50-b-PPhe40 | 55:45 | 65:35 | 5880 | 3675 |
| PLLA25-b-PPhe5 | 83:17 | 89:11 | 735 | 458 |
| PLLA25-b-PPhe13 | 65:35 | 79:21 | 1911 | 958 |
| PLLA10-b-PPhe25 | 28:72 | 24:76 | 3675 | 4617 |
| PLLA10-b-PPhe40 | 20:80 | 15:85 | 5880 | 8207 |




2.2. Physical Properties of Hybrid Copolymers



- (a)
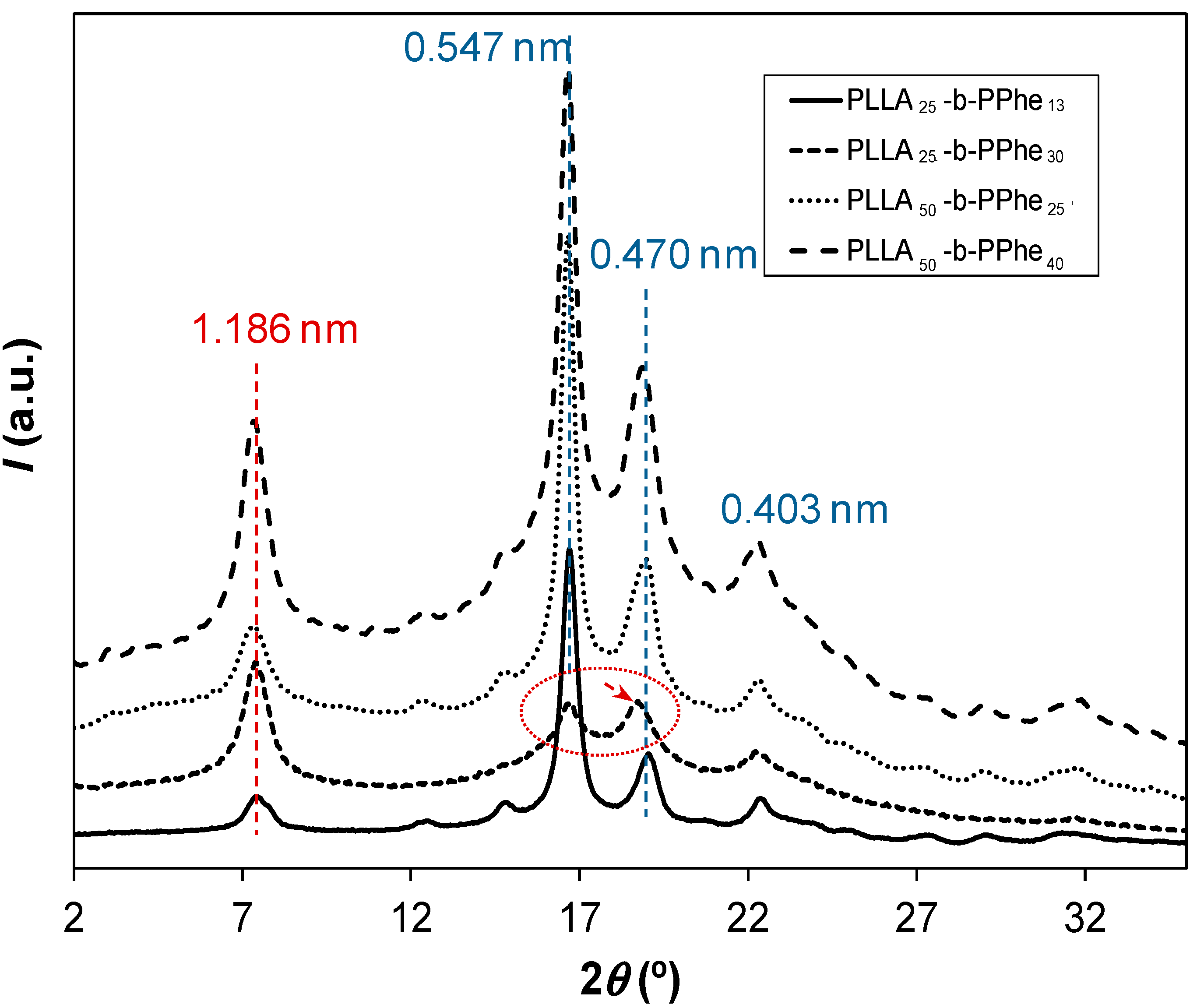
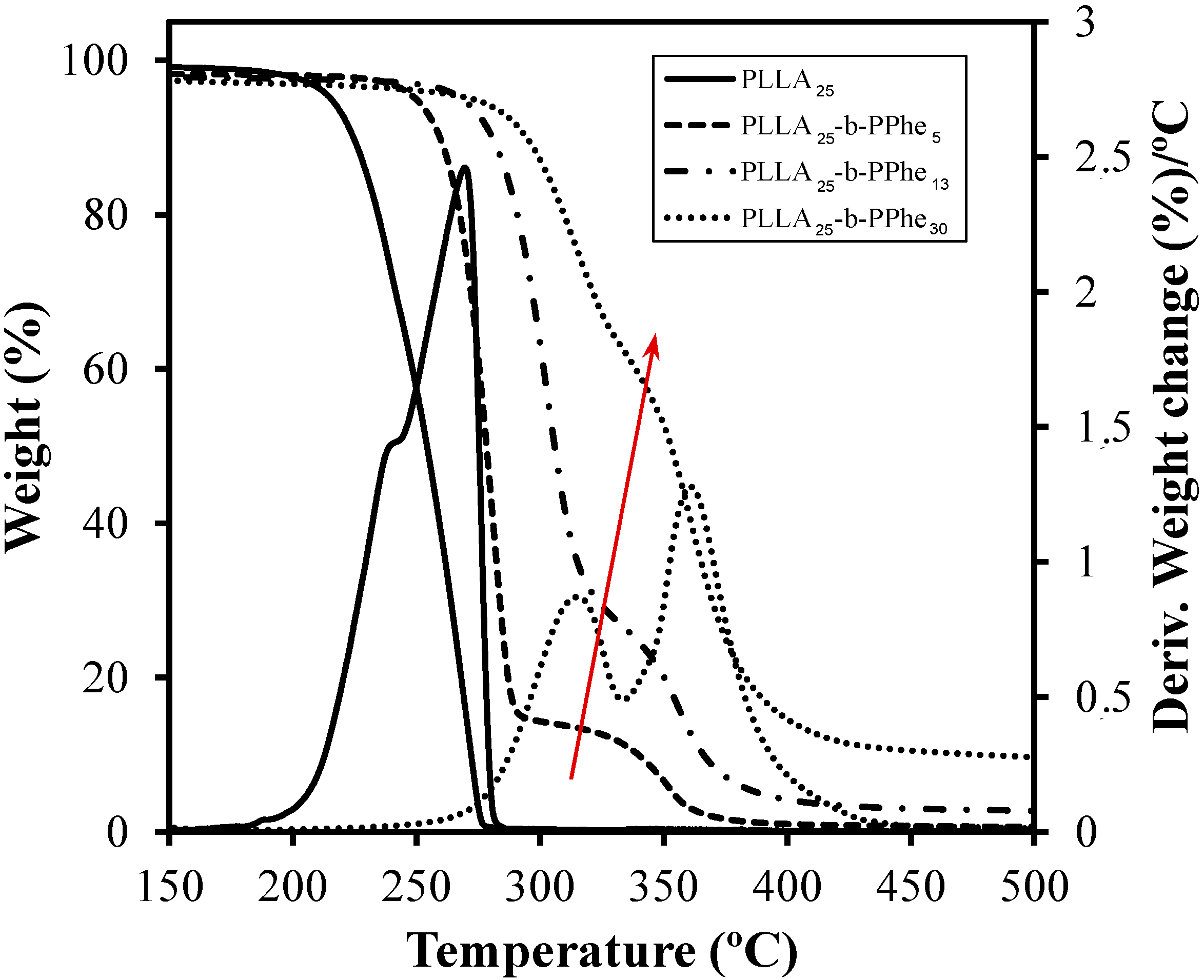
- Although peptide blocks gave rise to crystalline arrangements, only melting transitions associated with PLLA blocks were observed, as proved from X-ray diffraction data. Therefore, thermal decomposition seemed to occur before the melting temperature of PPhe was reached.
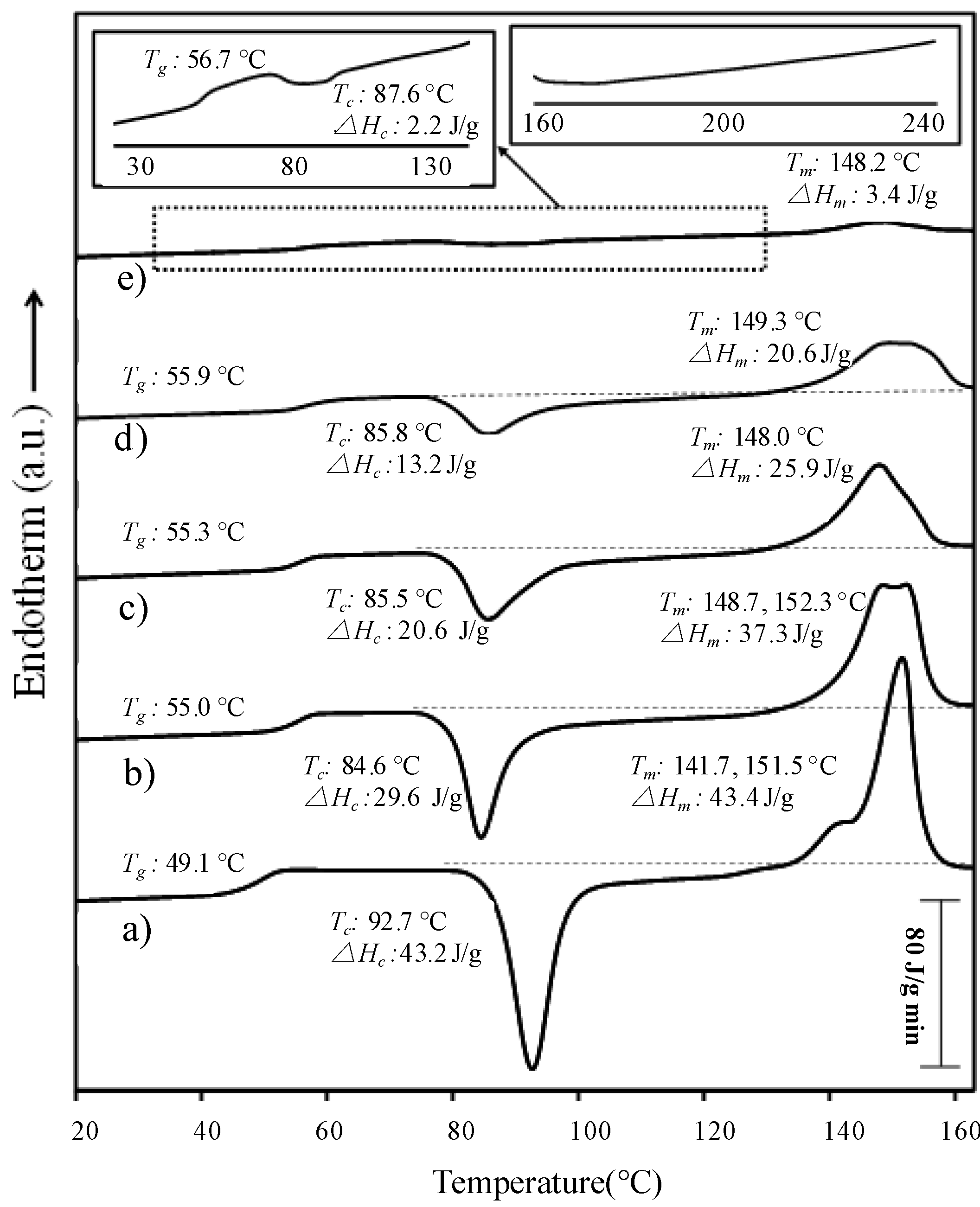
- (b)
- Melting of PLLA blocks was defined by two well differentiated peaks associated with a lamellar reorganization. The high temperature peak corresponds to recrystallized/reordered lamellae formed during the heating rate. The evolution observed for the copolymers indicated that this reorganization became less significant as the length of the PPhe block increased.
- (c)
- Glass transition temperature slightly increased with the PPhe content (i.e., from 49.1–56.7 °C for the PLLA25 series), indicating that some rigid l-Phe units were incorporated in the PLLA amorphous phase. No transition was detected for PPhe blocks, suggesting that they formed crystalline aggregates. Although an increase in the glass transition temperature may also be justified by the molecular weight increase caused by the incorporation of peptide blocks, the effect does not seem highly relevant when Tgs of PLLA50-b-PPhe25 and PLLA25-b-PPhe13 samples are compared (i.e., 55.9 versus 55.3 °C).
- (d)
- PLLA melting enthalpy logically decreased when the PPhe content in the sample increased. This reduction is higher than expected according to the copolymer composition (e.g., the melting enthalpy changed from 43.4–3.4 J/g when a block of 30 l-Phe units was incorporated in the PLLA25 block, while this enthalpy should be close to 20 J/g according to the real weight of PLLA in the copolymer). Therefore, the presence of peptide blocks hindered crystal growth of PLLA and the sample became more amorphous, as previously deduced from X-ray diffraction data.
- (e)
- PPhe blocks may act as effective nucleation agents since the cold crystallization peak observed for hybrid copolymers was shifted towards lower temperatures compared to that of the PLLA homopolymer. It is also significant that the difference between melting enthalpy and crystallization enthalpy of copolymers was higher than that found for PLLA homopolymers. This suggests an enhanced crystallization during the previous fast cooling run performed from temperatures higher than the PLLA melting temperature. Note that the crystalline PPhe domains were not melted to prevent degradation, and were therefore ideal nucleating agents.
- (f)
- Crystallinity slightly decreased with the molecular weight of the sample for a given composition (e.g., clear differences are found between DSC traces of PLLA50-b-PPhe25 and PLLA25-b-PPhe13 samples in Figure 11).
| Sample | Tg (°C) | Tc (°C) | ∆Hc (J/g) | Tm (°C)a | ∆Hm (J/g) |
|---|---|---|---|---|---|
| PLLA25-NH2 | 49.1 | 92.7 | 43.2 | 141.7, 151.5 | 43.4 |
| PLLA25-b-PPhe5 | 55.0 | 84.6 | 29.6 | 148.7, 152.3 | 37.3 |
| PLLA25-b-PPhe13 | 55.3 | 85.5 | 20.6 | 148.0 | 25.9 |
| PLLA25-b-PPhe30 | 56.7 | 87.6 | 2.2 | 148.2 | 3.4 |
| PLLA50-b-PPhe10 | 59.6 | 86.3 | 21.2 | 153.9 | 26.4 |
| PLLA50-b-PPhe25 | 55.9 | 85.8 | 13.2 | 149.3, 152.4 | 20.6 |
| PLLA50-b-PPhe40 | 54.1 | 86.3 | 4.1 | 152.8 | 5.7 |


3. Experimental
3.1. Materials
3.2. Polymerization of l-lactide
3.3. N-Boc Deprotection of Polylactide
3.4. Polimerization of l-Phe-NCA
3.5. Characterization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Morell, M.; Puiggalí, J. Hybrid Block Copolymers constituted by peptides and synthetic polymers: An overview of synthetic approaches, supramolecular behavior and potential applications. Polymers 2013, 5, 188–224. [Google Scholar] [CrossRef] [Green Version]
- Schlaad, H.; Antonietti, M. Block copolymers with amino acid sequences: Molecular chimeras of polypeptides and synthetic polymers. Eur. Phys. J. E 2003, 10, 17–23. [Google Scholar] [CrossRef]
- Klok, H.A.; Ayres, L. Peptide Hybrid Polymers; Klok, H.A., Schlaad, H., Eds.; Springer: Berlin, Germany, 2006. [Google Scholar]
- Börner, H.G. Strategies exploiting functions and self-assembly properties of bioconjugates for polymer and materials sciences. Prog. Polym. Sci. 2009, 34, 811–851. [Google Scholar] [CrossRef]
- Lutz, J.F.; Börner, H.G. Modern trends in polymer bioconjugates design. Prog. Polym. Sci. 2008, 33, 1–39. [Google Scholar] [CrossRef]
- Gallot, B. Comb-like and block liquid crystalline polymers for biological applications. Prog. Polym. Sci. 1996, 21, 1035–1088. [Google Scholar] [CrossRef]
- Langer, R.; Tirrell, D.A. Designing materials for biology and medicine. Nature 2004, 428, 487–491. [Google Scholar] [CrossRef]
- Nampoothiri, K.M.; Nair, N.R.; John, R.P. An overview of the recent developments in polylactide (PLA) research. Bioresour. Technol. 2010, 101, 8493–8501. [Google Scholar] [CrossRef]
- Caillol, S.; Lecommandoux, S.; Mingotaud, A.F.; Schappacher, M.; Soum, A.; Bryson, N.; Meyrueix, R. Synthesis and self-assembly properties of peptide-polylactide block copolymers. Macromolecules 2003, 36, 1118–1124. [Google Scholar] [CrossRef]
- Deng, C.; Rong, G.; Tian, H.; Tang, Z.; Chen, X.; Jing, X. Synthesis and characterization of poly(ethylene glycol)-b-poly(l-lactide)-b-poly(l-glutamic acid) triblock copolymer. Polymer 2005, 46, 653–659. [Google Scholar] [CrossRef]
- Dorresteijn, R.; Ragg, R.; Rago, G.; Billecke, N.; Bonn, M.; Parekh, S.H.; Battagliarin, G.; Peneva, K.; Wagner, M.; Klapper, M.; et al. Biocompatible polylactide-block-polypeptide-block-polylactide nanocarrier. Biomacromolecules 2013, 14, 1572–1577. [Google Scholar] [CrossRef]
- Deng, C.; Chen, X.; Sun, J.; Lu, T.; Wang, W.; Jing, X. RGD peptide grafted biodegradable amphiphilic triblock copolymer Poly(glutamic acid)-b-poly(l-lactide)-b-poly(glutamic acid): Synthesis and self-assembly. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 3218–3230. [Google Scholar] [CrossRef]
- Ju, M.; Gong, F.; Cheng, S.; Gao, Y. Fast and convenient synthesis of amine-terminated polylactide as a macroinitiator for ω-benzyloxycarbonyl-l-lysine-n-carboxyanhydrides. Int. J. Polym. Sci. 2011. [Google Scholar] [CrossRef]
- Sun, J.; Chen, X.; Lu, T.; Liu, S.; Tian, H.; Guo, Z.; Jing, X. Formation of reversible shell cross-linked micelles from the biodegradable amphiphilic diblock copolymer Poly(l-cysteine)-block-poly(l-lactide). Langmuir 2008, 24, 10099–10106. [Google Scholar] [CrossRef]
- Cho, Y.W.; Lee, J.; Lee, S.C.; Huh, K.M.; Park, K. Hydrotropic agents for study of in vitro paclitaxel release from polymeric micelles. J. Control. Release 2004, 97, 249–257. [Google Scholar] [CrossRef]
- Cao, H.; Yao, J.; Shao, Z. Synthesis of pol(γ-benzyl-l-glutamate) with well-defined terminals structures and its block polypeptides with alanine, leucine and phenylalanine. Polym. Int. 2012, 61, 774–779. [Google Scholar] [CrossRef]
- Waters, M.L. Aromatic interactions in model systems. Curr. Opin. Chem. Biol. 2002, 6, 736–741. [Google Scholar] [CrossRef]
- Gazit, E. A possible role for π-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. [Google Scholar]
- Reches, M.; Gazit, E. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science 2003, 300, 625–627. [Google Scholar] [CrossRef]
- Song, Y.; Challa, S.R.; Medforth, C.J.; Qiu, Y.; Watt, R.K.; Pena, D.; Miller, J.E.; Swol, F.V.; Shelnutt, J.A. Synthesis of peptide-nanotube platinum-nanoparticle composites. Chem. Commun. 2004, 7, 1044–1045. [Google Scholar]
- Korolkov, V.V.; Allen, S.; Roberts, C.J.; Tendler, S.J.B. Surface mediated l-phenylalanyl-l-phenylalanine assembly into large dendritic structures. Faraday Discuss. 2013, 166, 331–348. [Google Scholar]
- Murase, S.K.; Haspel, N.; del Valle, L.J.; Nussinov, R.; Puiggalí, J.; Alemán, C. Molecular characterization of l-phenylalanine terminated poly(l-lactide) conjugates. RSC Adv. 2014, 4, 23231–23241. [Google Scholar]
- Deming, T.J.; Cheng, J. Synthesis of polypeptides by ring-opening polymerization of α-amino acid N-carboxyanhydrides. Top. Curr. Chem. 2012, 310, 1–26. [Google Scholar]
- Gotsche, M.; Keul, H.; Höcke, H. Amino-termined poly (l-lactide)s as initiators for the polymerization of N-carboxyanhydrides: synthesis of poly(l-lactide)-block-poly(α-amino acid)s. Macromol. Chem. Phys. 1995, 196, 3891–3903. [Google Scholar]
- Yuan, M.; Deng, X. Synthesis and characterization of poly(ethylene glycol)-block-poly(amino acid) copolymer. Eur. Polym. J. 2001, 37, 1907–1912. [Google Scholar] [CrossRef]
- Sacristán, M.; Plantá, X.; Morell, M.; Puiggalí, J. Effects of ultrasonic vibration on the micro-molding processing of polylactide. Ultrason. Sonochem. 2013, 21, 376–386. [Google Scholar]
- McNeil, I.C.; Leiper, H.A. Degradation studies of some polyesters and polycarbonates—1. Polylactide: General features of the degradation under programmed heating conditions. Polym. Degrad. Stab. 1985, 11, 267–285. [Google Scholar]
- Hoogenboom, R.; Wouters, D.; Schubert, U.S. l-lactide polymerization utilizing a hydroxy-functionalized 3,6-bis(2-pyridyl)pyridazine as supramolecular (co)initiator: Construction of polymeric [2 × 2] grids. Macromolecules 2003, 36, 4743–4749. [Google Scholar] [CrossRef]
- De Santis, P.; Kovacs, A.J. Molecular conformation of poly(S-lactic acid). Biopolymers 1968, 6, 299–306. [Google Scholar] [CrossRef]
- Hoogsteen, W.; Postema, A.R.; Pennings, A.J.; Brinke, G.T.; Zugenmaier, P. Crystal structure, conformation, and morphology of solution-spun poly(l-lactide) fibers. Macromolecules 1990, 23, 634–642. [Google Scholar] [CrossRef]
- Miyata, T.; Masuko, T. Morphology of poly(l-lactide) solution-grow. Polymer 1997, 38, 4003–4009. [Google Scholar] [CrossRef]
- Kobayashi, J.; Asahi, T.; Ichiki, M.; Okikawa, A.; Suzuki, H.; Watanabe, T.; Fukada, E.; Shikinami, Y. Structural and optical properties of poly lactic acids. J. Appl. Phys. 1995, 77, 2957–2973. [Google Scholar] [CrossRef]
- Eling, B.; Gogolewski, S.; Pennings, A.J. Biodegradable materials of poly(l-lactic Acid). 1. Melt spun and solution spun fibres. Polymer 1982, 23, 1587–1593. [Google Scholar]
- Puiggali, J.; Ikada, Y.; Tsuji, H.; Cartier, L.; Okihara, T.; Lotz, B. The frustrated structure of poly(l-lactide). Polymer 2000, 41, 8921–8930. [Google Scholar] [CrossRef]
- Lotz, B.; Brack, A. The use of polypeptides as models in fibrous protein structure investigations. In Applied Fibre Science; Happey, F., Ed.; Academic Press: London, UK, 1979; Volume 3, pp. 371–410. [Google Scholar]
- Kopinke, F.D.; Remmler, M.; Mackenzie, K.; Möder, M.; Wachsen, O. Thermal decomposition of biodegradable polyesters—II. Poly(lactic acid). Polym. Degrad. Stab. 1996, 53, 329–342. [Google Scholar] [CrossRef]
- Márquez, Y.; Franco, L.; Puiggalí, J. Thermal degradation studies of poly(trimethylene carbonate) blends with either polylactide or polycaprolactone. Thermochim. Acta 2014, 585, 71–80. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Planellas, M.; Puiggalí, J. Synthesis and Properties of Poly(l-lactide)-b-poly (l-phenylalanine) Hybrid Copolymers. Int. J. Mol. Sci. 2014, 15, 13247-13266. https://doi.org/10.3390/ijms150813247
Planellas M, Puiggalí J. Synthesis and Properties of Poly(l-lactide)-b-poly (l-phenylalanine) Hybrid Copolymers. International Journal of Molecular Sciences. 2014; 15(8):13247-13266. https://doi.org/10.3390/ijms150813247
Chicago/Turabian StylePlanellas, Marc, and Jordi Puiggalí. 2014. "Synthesis and Properties of Poly(l-lactide)-b-poly (l-phenylalanine) Hybrid Copolymers" International Journal of Molecular Sciences 15, no. 8: 13247-13266. https://doi.org/10.3390/ijms150813247
APA StylePlanellas, M., & Puiggalí, J. (2014). Synthesis and Properties of Poly(l-lactide)-b-poly (l-phenylalanine) Hybrid Copolymers. International Journal of Molecular Sciences, 15(8), 13247-13266. https://doi.org/10.3390/ijms150813247