1. Introduction
Cancer is the leading cause of deaths worldwide, and thus a seriously threat to human health. Due to the complexity of tumorigenesis and difficulty of cancer therapy, combinations of chemotherapeutic drugs with other treatment modalities like nucleic acid have shown great promise in cancer therapy [
1,
2]. Paclitaxel (PTX) was an effective chemotherapeutic drug and widely used for cancer treatment, including melanoma, ovarian cancer and hepatoma [
3,
4]. However, more and more cancers have shown decreased response to the mono-chemotherapy of PTX [
5,
6], thus, it is necessary to combine gene therapy with PTX to achieve synergistic combination therapeutic effect.
However, the drug and gene leakage in blood circulation, the non-specific distribution and the immune response to normal tissue seriously limited the clinical application of drug/gene combination treatment [
7]. Therefore, it is necessary to design a rational multifunctional nanocarrier to co-deliver PTX and DNA, overcoming the above mentioned problems. Cationic liposomes were reported widely in gene delivery [
8,
9], and the multilamellar liposomes were expected to show advantages in co-delivering the hydrophobic PTX and the hydrophilic DNA. Considering polyethylenimine (PEI) was one of the most effective nonviral gene delivery polymers [
10,
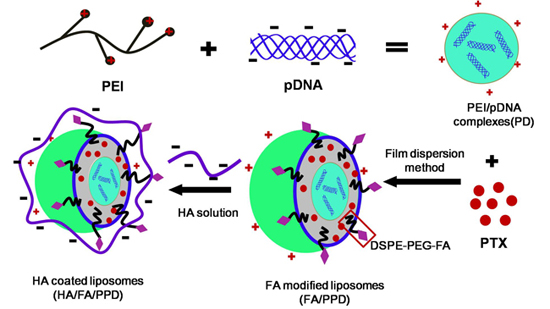
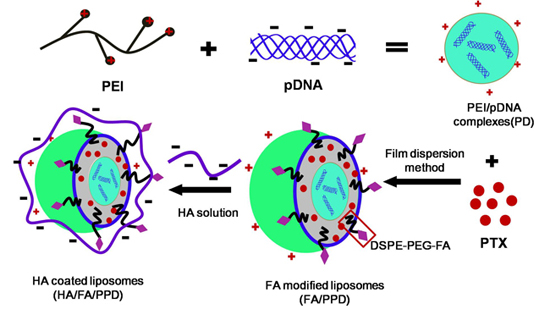
11], the condensed PEI/DNA complexes were chosen as the cationic core, to prepare the DNA and PTX co-loaded cationic liposomes (PPD) (as shown in
Scheme 1).
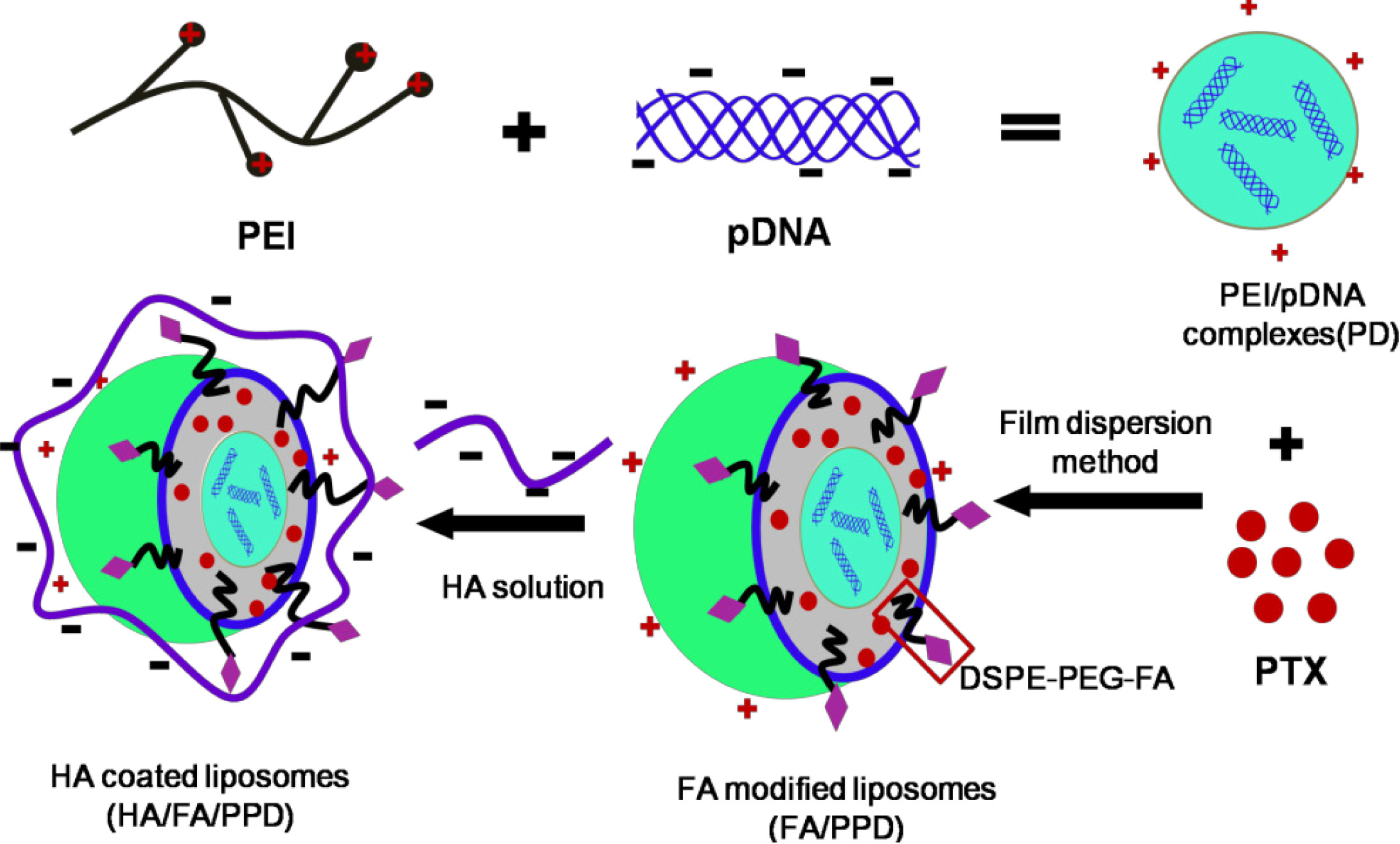
Scheme 1.
Schematic illustration of formation of HA/FA/PPD (hyaluronic acid (HA) and folate (FA)-modified liposomes). Firstly, PEI (polyethylenimine) bind DNA to form the condensed cationic PEI/DNA complexes, which were chosen as the cationic core of the liposomes. Subsequently, PTX (paclitaxel) and PEI/DNA complexes were co-loaded in the DSPE-PEG2000-FA-modified liposomes, forming the FA-modified cationic liposomes (FA/PPD). Lastly, the cationic FA/PPD was added to the anionic HA solution to obtain the HA coated FA/PPD (HA/FA/PPD) by electrostatic attraction.
Scheme 1.
Schematic illustration of formation of HA/FA/PPD (hyaluronic acid (HA) and folate (FA)-modified liposomes). Firstly, PEI (polyethylenimine) bind DNA to form the condensed cationic PEI/DNA complexes, which were chosen as the cationic core of the liposomes. Subsequently, PTX (paclitaxel) and PEI/DNA complexes were co-loaded in the DSPE-PEG2000-FA-modified liposomes, forming the FA-modified cationic liposomes (FA/PPD). Lastly, the cationic FA/PPD was added to the anionic HA solution to obtain the HA coated FA/PPD (HA/FA/PPD) by electrostatic attraction.
In order to achieve efficient drug/gene delivery into target cells, the most promising strategies involved the application of various targeting moieties or ligands that bind specifically to receptors overexpressed on malignant cells [
12,
13]. Folate (FA), a nontoxic low-weight compound with low immunogenicity, is vital for tumor cell proliferation and survival [
14]. The folate receptor (FR) is overexpressed in a wide range of human cancer cells, including ovarian cancer, melanoma, head and neck cancers, presenting an effective means of selectively delivering drug/gene to tumors [
15,
16]. Therefore, FA was suitable to modify PPD (FA/PPD).
The cationic property of FA/PPD allows it to easily interact with serum complexes compounds, forming aggregates, which are subjected to
in vivo phagocyte capture, giving rise to vector degradation in turn associated with PTX/DNA release and degradation [
17]. Hyaluronic acid (HA), an endogenous component of extracellular matrix, is a negatively charged linear polysaccharide [
18,
19]. Thus, the negative HA could coat the surface of FA/PPD by electrostatic attraction (HA/FA/PPD), resulting in a biomimetic and anionic layer and subsequently preventing liposomes from aggregating. In
in vivo circulation, HA coated liposomes could bind specifically to CD44, which is overexpressed in cancer cells [
18,
19]. In addition, the outer HA layer would inevitably suffer from an enzyme degradation, which would subsequently expose the FA moiety and target to cancer cells again. Consequently, the HA-coated and FA-modified HA/FA/PPD was expected to achieve dual targeting co-delivery of PTX and DNA.
In the present study, the plasmid pCMV-EGFP (pEGFP-N1) carrying enhanced green fluorescent protein (EGFP) were chosen as model gene. HA- and FA-modified dual targeting biomimetic liposomes were prepared by film dispersion method to co-deliver PTX and DNA (HA/FA/PPD), expecting to target to tumor sites, enhance transfection efficiency and realize the optimal combination therapy (as shown in
Scheme 1). FA/PPD and PPD were simultaneously prepared to serve as controls. The cytotoxicity of HA/FA/PD (PTX-free HA/FA/PPD) and FA/PD (PTX-free FA/PPD) were studied by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay on murine malignant melanoma cell line (B16) and human hepatocellular carcinoma cell line (HepG2), to evaluate the safety of the nanovectors. The release profile of PTX from HA/FA/PPD was also evaluated by dynamic dialysis method. The stability of HA/FA/PPD and FA/PPD in presence of plasma and the ability to protect DNA against DNase I were investigated to testify the protection function of biomimetic HA layer of HA/FA/PPD. The transfection efficiency and the targeted cellular uptake of HA/FA/PPD, FA/PPD and PPD were evaluated on CD44-positive and FR-positive B16 cells by fluorescence microscope and flow cytometry, respectively [
20,
21], while the co-delivery efficiency of PTX and pDNA into the same tumor cell was evaluated on CD44-positive and FR-negative HepG2 cells [
22,
23].
3. Experimental Section
3.1. Materials
Paclitaxel (PTX), rhodamine labeled paclitaxel (Rhodamine-Paclitaxel, Rho-PTX), cholesterol (Chol) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were obtained from Sigma-Aldrich (St. Louis, MS, USA). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-folate poly-(ethylene glycol)2000 (DSPE-PEG2000-FA), dioleoylphosphatidylethanolamine (DOPE) and egg l-α-phosphatidylcholine (ePC) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Branched poly(ethyleneimine) (PEI, MW 25 kDa) was purchased from Sigma-Aldrich (USA). The plasmid pCMV-EGFP (pEGFP-N1) carrying enhanced green fluorescent protein (EGFP) under cytomegalovirus (CMV) promoter was propagated in Escherichia coli and purified by Endo Free Plasmid Maxi Kit (Qiagen, Hilden, Germany). Hyaluronic acid (HA, 300 kDa) was provided by Shandong Freda Biochem Co., Ltd. (Jinan, China). Agarose was purchased from Biowest (Nuaille, France). DNase I enzyme and goldview was purchased from Solarbio (Shanghai, China). Lipofectamine™ 2000 was purchased from Invitrogen by Life Technologies (Carlsbad, CA, USA). All other reagents were of commercial special grade and used without further purification.
3.2. Cell Lines and Cell Culture
Murine melanoma cell line B16 and human hepatoma HepG2 cells were purchased from the Chinese Academy of Sciences (Shanghai, China) and cultured in RPMI-1640 media at 37 °C under 5% CO2. All the media were supplemented with 10% (v/v) fetal bovine serum from Sijiqing Co., Ltd. (Hangzhou, China), streptomycin at 100 μg/mL and penicillin at 100 U/mL.
3.3. Preparation of PEI/DNA Complexes
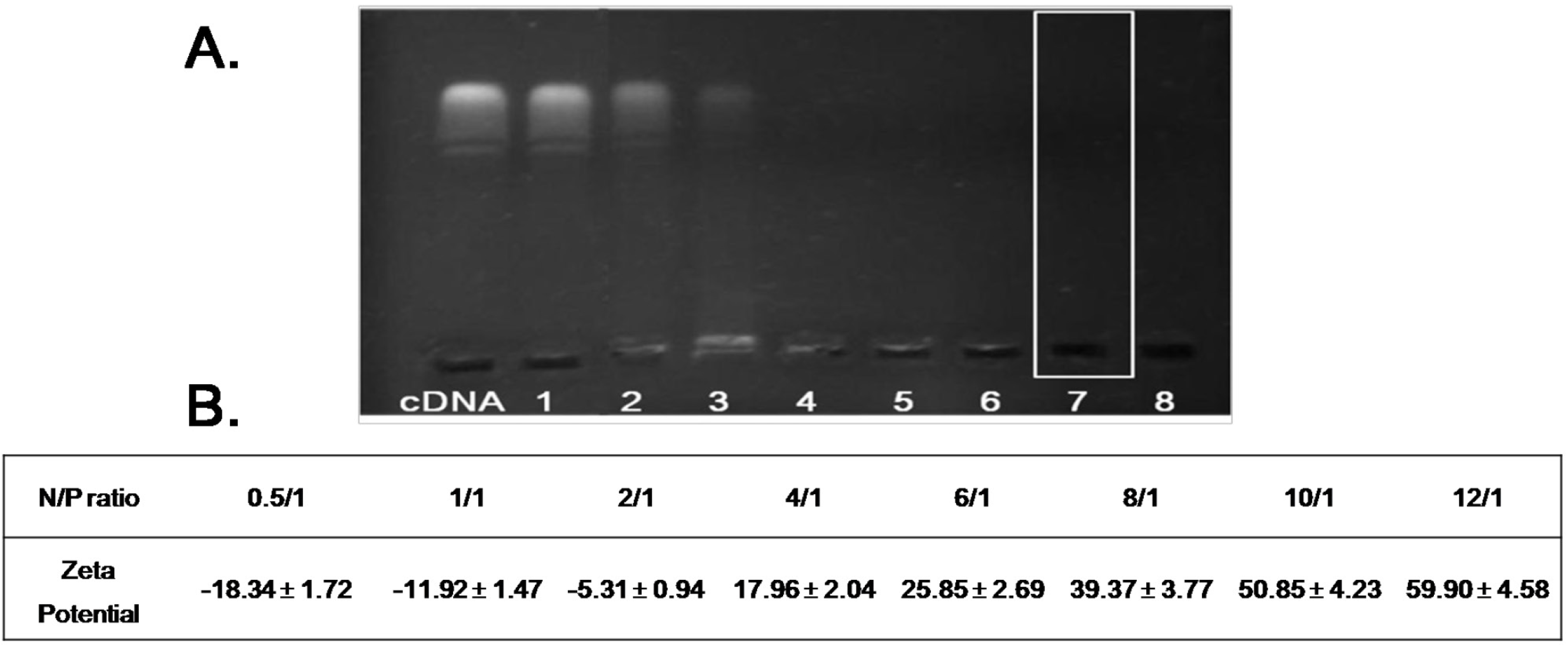
Stock PEI solution (1000 μg/mL) and DNA solution (100 μg/mL) were prepared before each experiment and stocked in hydroxyethyl piperazine ethanesulfonic acid (HEPES)-buffered saline (HBS, pH 7.4). The PEI/DNA complex was prepared at various molar ratios of PEI nitrogen (N) to DNA phosphate (P) up to N/P = 12. Specifically, the PEI/DNA complexes were prepared by slowly mixing the solution of DNA in the PEI solution at N/P ratios of 0.5/1, 1/1, 2/1, 4/1, 6/1, 8/1, 10/1 and 12/1, respectively. Samples were continuously stirred during addition and equilibrated at room temperature for 20 min before measurement. Complexes were freshly prepared before each individual measurement. A gel retardation assay was used to determine the stability of the PEI/DNA complexes. Each well of the gels was loaded with PEI/DNA complexes containing 1 μg of plasmid DNA while the naked DNA served as control. After loading the complexes onto the agarose gel, electrophoresis was carried out in constant voltage mode at 90 V for 20 min.
3.4. Preparation of Paclitaxel and PEI-DNA Co-Loaded Liposomes (PPD)
Liposomes were prepared using a slight modification of the previously described method for preparing multi-lamellar vesicles [
26]. To prepare empty liposomes, ePC, DOPE and were dissolved in chloroform at a mass ratio of 7:4:2. While PTX-loaded liposomes were being prepared, PTX-chloroform stock solution (1 mg/mL) was combined with the mixed lipids at a mass ratio of 7:4:2:0.8. (ePC:DOPE:Chol:PTX). The mixture was evaporated to dryness in a round-bottomed flask using a rotary evaporator at room temperature. The resulting thin lipid film was dried using nitrogen for an additional 10 min to evaporate any dehydrated chloroform. To prepare PTX/PEI-DNA co-loaded liposomes, 2 mL of PEI/DNA complexes prepared in
Section 3.2 was slowly added to the round-bottomed flask and then gently vibrated to facilitate hydration of the thin lipid film. The resulting multi-lamellar liposomes were then sonicated using a Sonic Dismembrator (Model 500; Thermo Fisher Scientific, Pittsburgh, PA, USA) for 3 min at room temperature to form homogenized cationic PPD.
3.5. Preparation of FA Modified Paclitaxel and DNA Loaded Liposomes (FA/PPD)
To prepare FA modified PPD, the covalent lipid DSPE-PEG-FA was added to the lipid mixture (5%, w/w), making the final mass ratio of ePC:DOPE:Chol:DSPE-PEG-FA 7:4:2:0.65. After that, the FA/PPD was also prepared by the modified film dispersion method and details as shown in 3.4.
3.6. Preparation of HA-Coated Targeting Liposomes (HA/FA/PPD)
The HA coated FA/PPD was prepared by electrostatic attraction. Briefly, 1 mL of FA/PPD dispersion was slowly added under vigorous stirring to HA solution (0.5 mg/mL, pH 6.0) at mass ratio of 0:1, 0.5:1, 1:1, 2:1, 3:1 and 4:1 (HA: FA/PPD, w/w). After incubation for 20 min at room temperature, the HA-coated FA/PPD (HA/FA/PPD) was finally obtained.
3.7. Characterization of FA/PPD and HA/FA/PPD
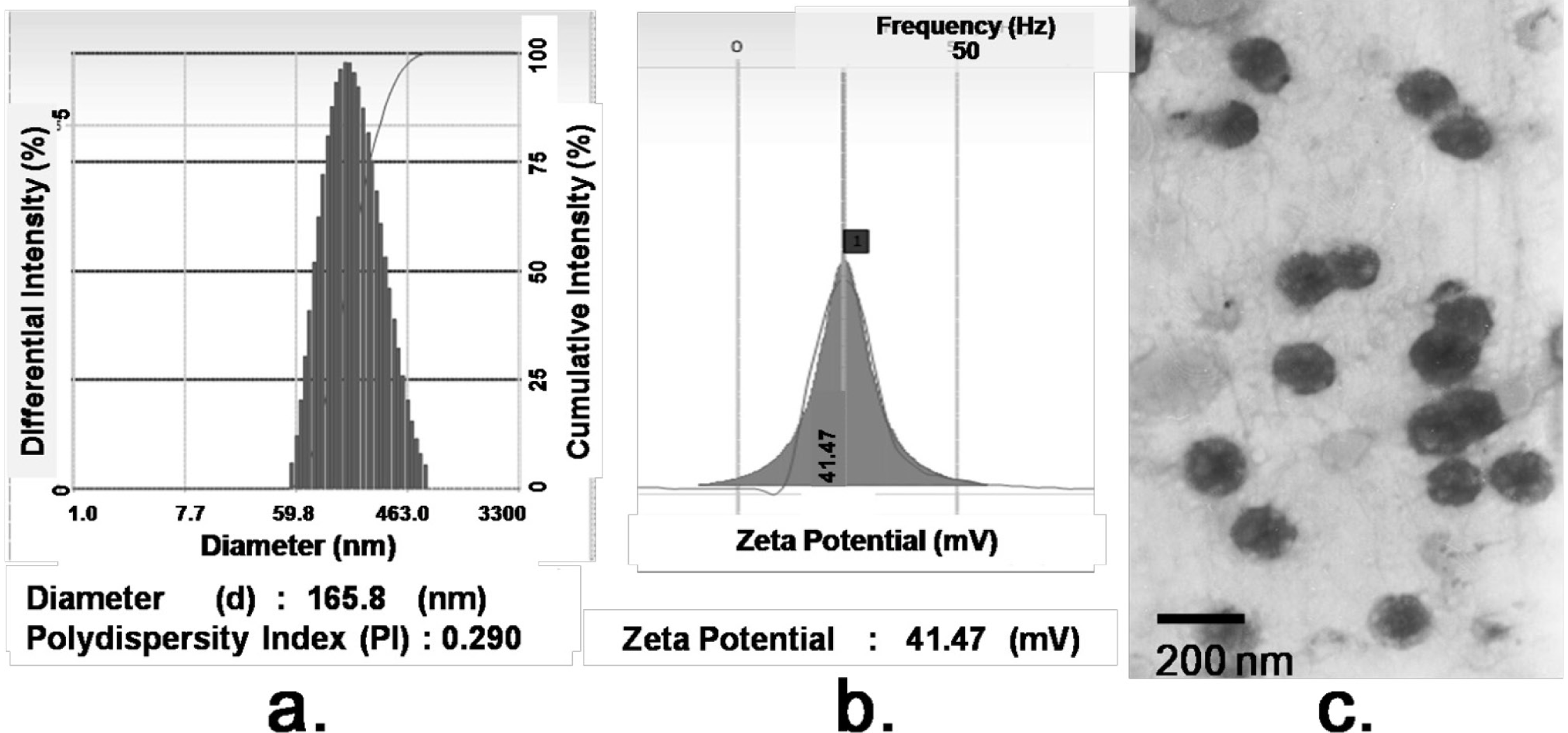
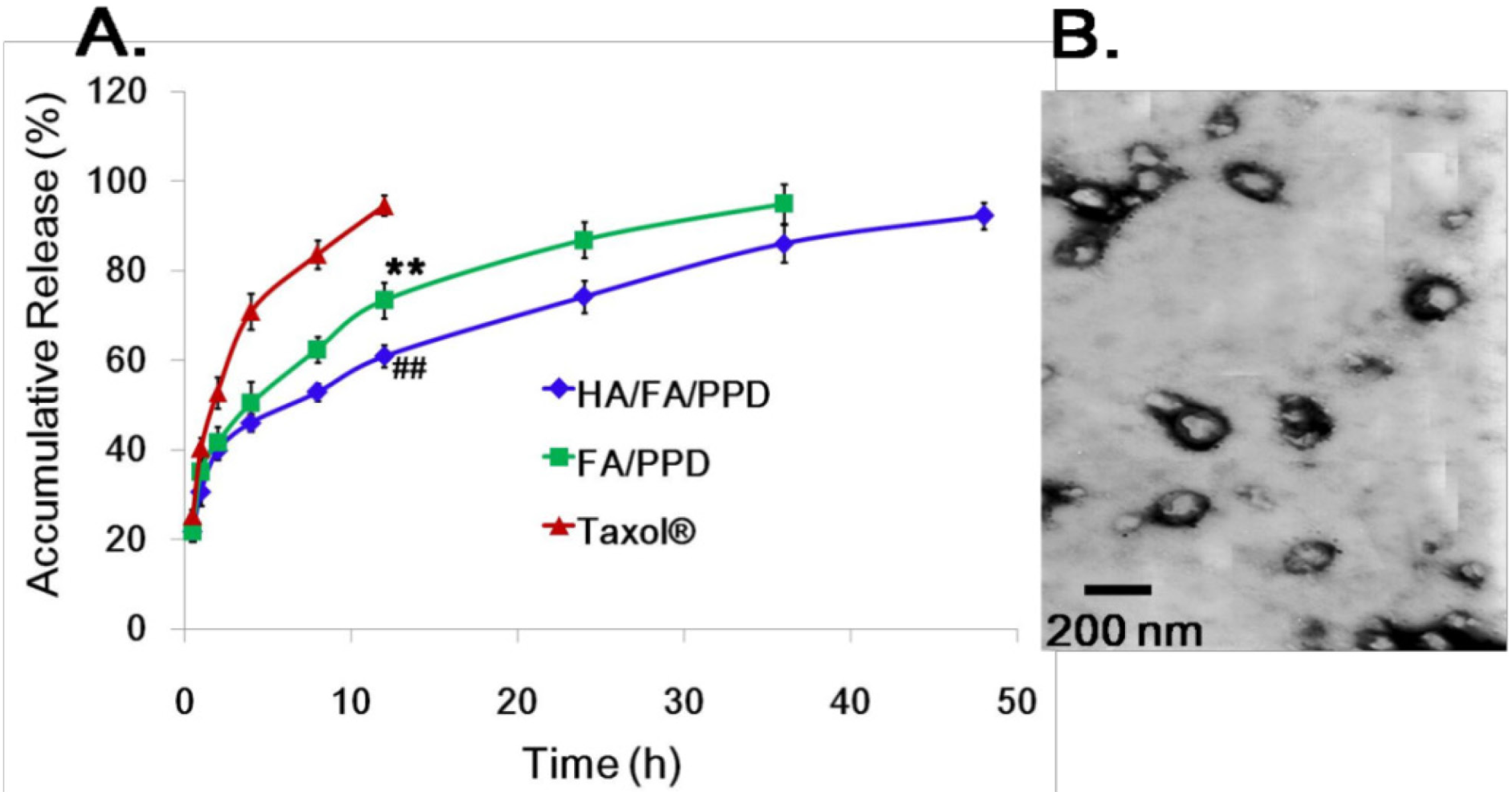
The morphology of FA/PPD and HA/FA/PPD was investigated by transmission electron microscopy (TEM) (JEM-1200EX, JEOL Ltd., Tokyo, Japan). For TEM studies, the FA/PPD and HA/FA/PPD dispersion were dropped on the surface of copper grid, and then 2% aqueous solution of sodium phosphotungstate was added for negative staining. After air-drying, the copper grid was placed to TEM and investigated. The particle size and zeta potential of FA/PPD and HA/FA/PPD were measured by laser light scattering using Zetasizer 3000SH (Malvern Instruments Ltd., Malvern, UK). All measurements were carried out in triplicate.
3.8. Stability of HA/FA/PPD in the Presence of Plasma
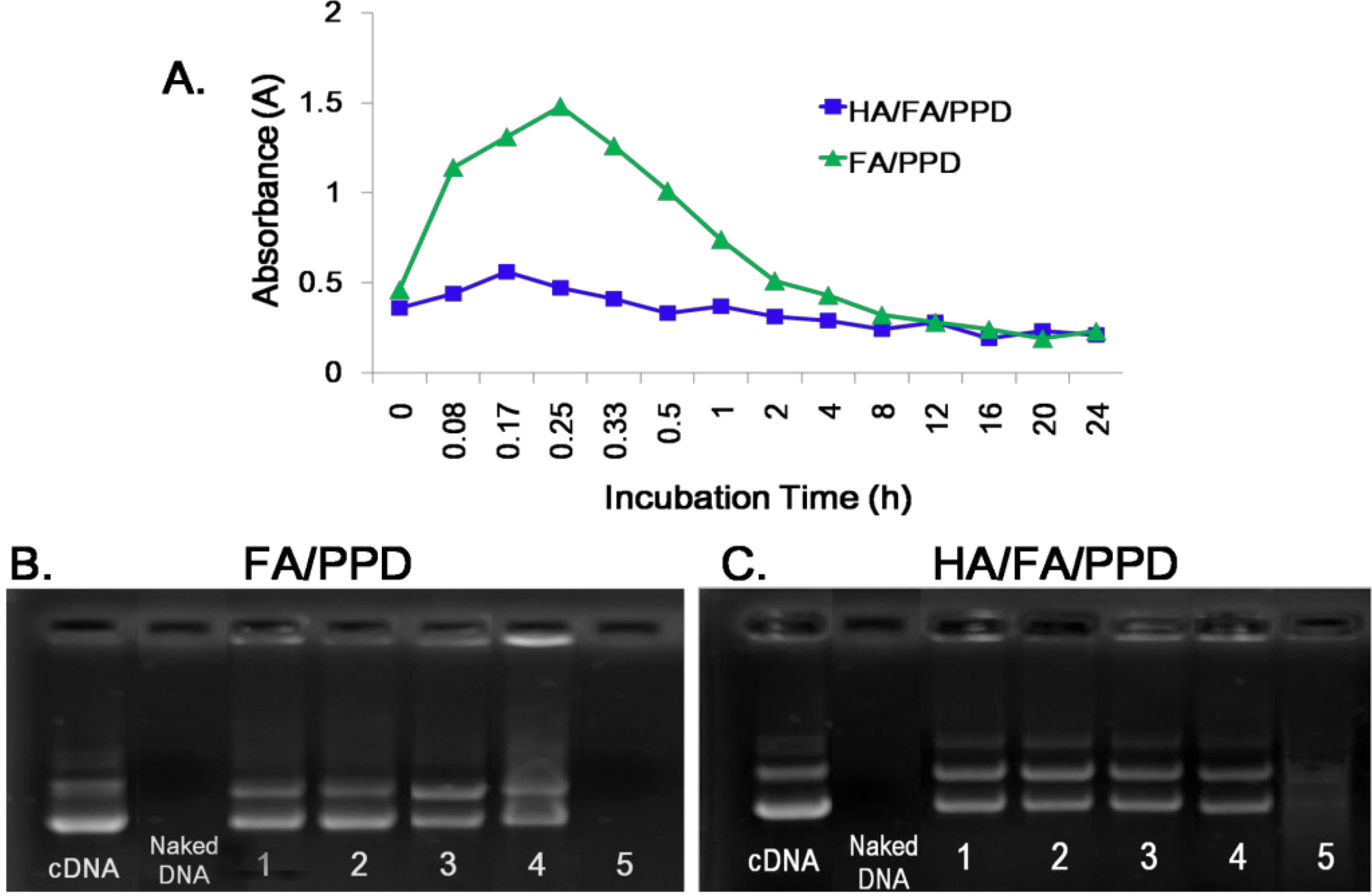
To estimate the stability of HA/FA/PPD under physiologically relevant conditions, the turbidity change of HA/FA/PPD suspension in the presence of plasma was evaluated. Specifically, a freshly prepared HA/FA/PPD solution (200 µL) was added to 1 mL of 50% plasma at pH 7.4 and the mixed solution was incubated at 37 °C with mild stirring. The turbidity of the mixed solution was measured by an ultraviolet-visible spectrophotometer (UV-2102PC; Unico Ltd., Dayton, NJ, USA) at 450 nm after 0.08, 0.17, 0.25, 0.33, 0.5, 1, 2, 4, 5, 12, 16, 20 and 24 h of incubation, respectively. FA/PPD with same treatment served as control.
3.9. DNase I Protection Assay
To test whether HA/FA/PPD and FA/PPD could protect loaded DNA from nuclease degradation, DNase I-mediated degradation was evaluated using agarose gel electrophoresis. In brief, 20 µL of HA/FA/PPD and FA/PPD containing 1 µg of DNA was incubated with DNase I (0.2 U/µg DNA) in DNase I buffer. The suspensions were incubated in a shaking water bath at 37 °C and 100 rpm for 0.5, 1, 2, 3 and 4 h, respectively. After incubation, ethylenediaminetetraacetic acid solution (0.5 M, pH 8.0) was added to terminate the enzymatic degradation reaction. To separate DNA from HA/FA/PPD and FA/PPD, TE buffer containing 1% heparin was added to the suspensions. The suspensions were then placed in a 37 °C shaking water bath at 100 rpm for another 2 h. After that, the fluorescent intensity of bands corresponding to DNA and their electrophoretic mobility were analyzed by gel electrophoresis. Naked DNA (1 µg) incubated with DNase I for 30 min served as control.
3.10. In Vitro Release Studies
The
in vitro release of PTX from HA/FA/PPD and FA/PPD was evaluated by dynamic dialysis method [
27]. Briefly, HA/FA/PPD and FA/PPD suspensions were diluted with de-ionized water to final PTX concentration of 100 µg/mL and placed into a pre-swelled dialysis bag with 8–12 kDa molecular weight cutoff. The dialysis bag was incubated in 15 mL release media (PBS containing 0.5% of Tween-80) at 37 ± 0.5 °C with stirring at 100 rpm. At predetermined time intervals, 1 mL solution was taken out and 15 mL fresh media was filled to replace the remaining release media. The amount of PTX in samples was then determined by high performance liquid chromatography (HPLC) method.
3.11. In Vitro Cytotoxicity Studies
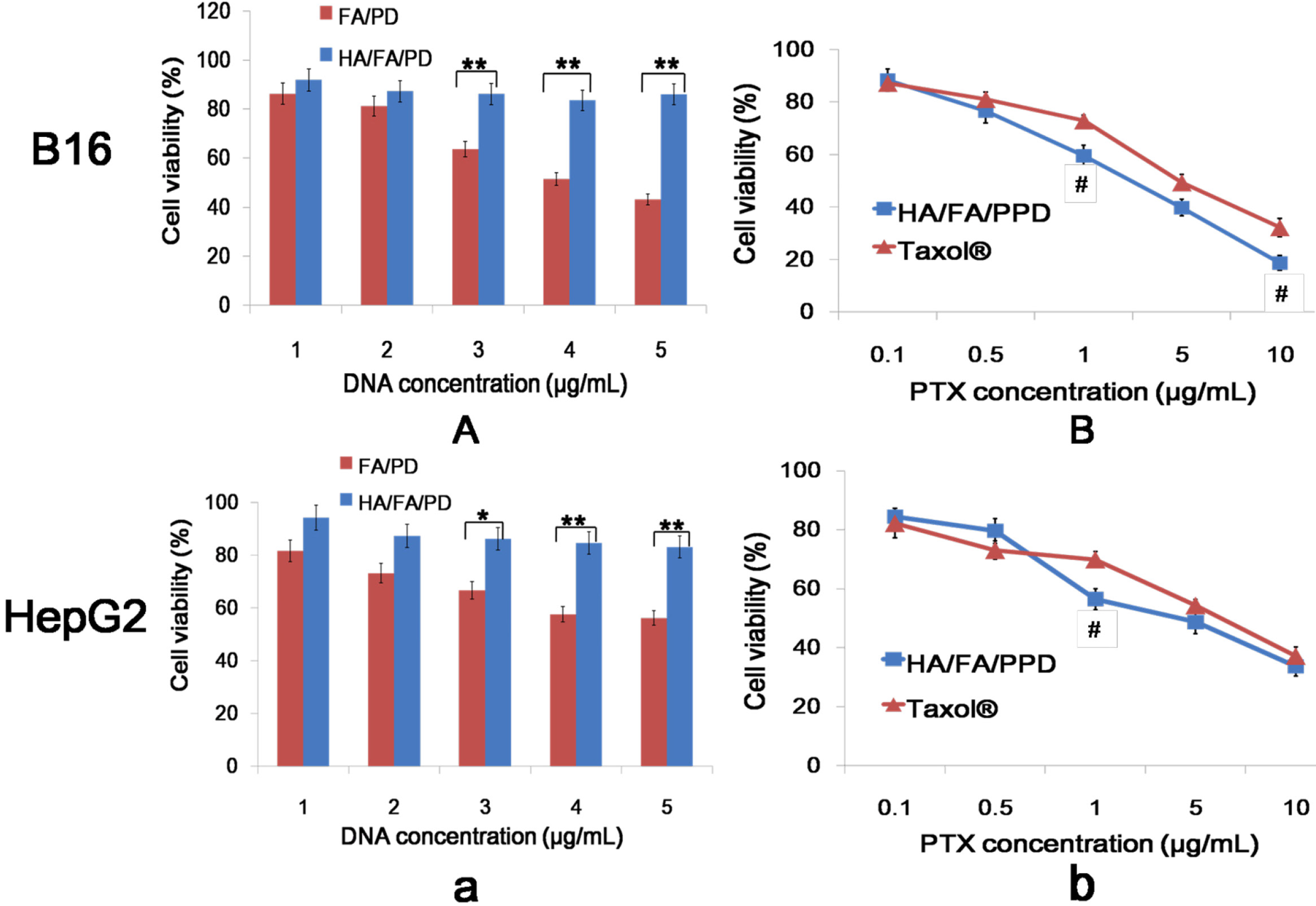
Murine melanoma cell line B16 and human hepatoma HepG2 cells were selected to investigate the in vitro cytotoxicity study. The in vitro cytotoxicity of HA/FA/PPD and FA/PPD on B16 and HepG2 cells were investigated by MTT assay. All concentrations were expressed in PTX equivalents. In brief, cells were seeded into a 96-well plate at a density of 4000 cells/well. After 48 h incubation with a series of doses of Taxol®, HA/FA/PPD and FA/PPD (final PTX concentration were 0.1, 0.5, 1, 5 and 10 μg/mL, respectively), 20 μL of MTT solution in PBS (5 mg/mL) was added to each well. After another 4 h, the incubation media was removed and 200 μL of DMSO was subsequently added to each well to dissolve formazan crystals, and measured at 570 nm. Moreover, to evaluate the safety of the prepared nanovector, the FA/PD (PTX free FA/PPD) and the HA/FA/PD (PTX free HA/FA/PPD) were prepared with the abovementioned method and the cytotoxicity at different DNA concentrations (1, 2, 3, 4, and 5 μg/mL) were investigated by abovementioned method. All experiments were repeated independently thrice and the relative cell viability (%) compared to control cells was calculated.
3.12. In Vitro Transfection Study
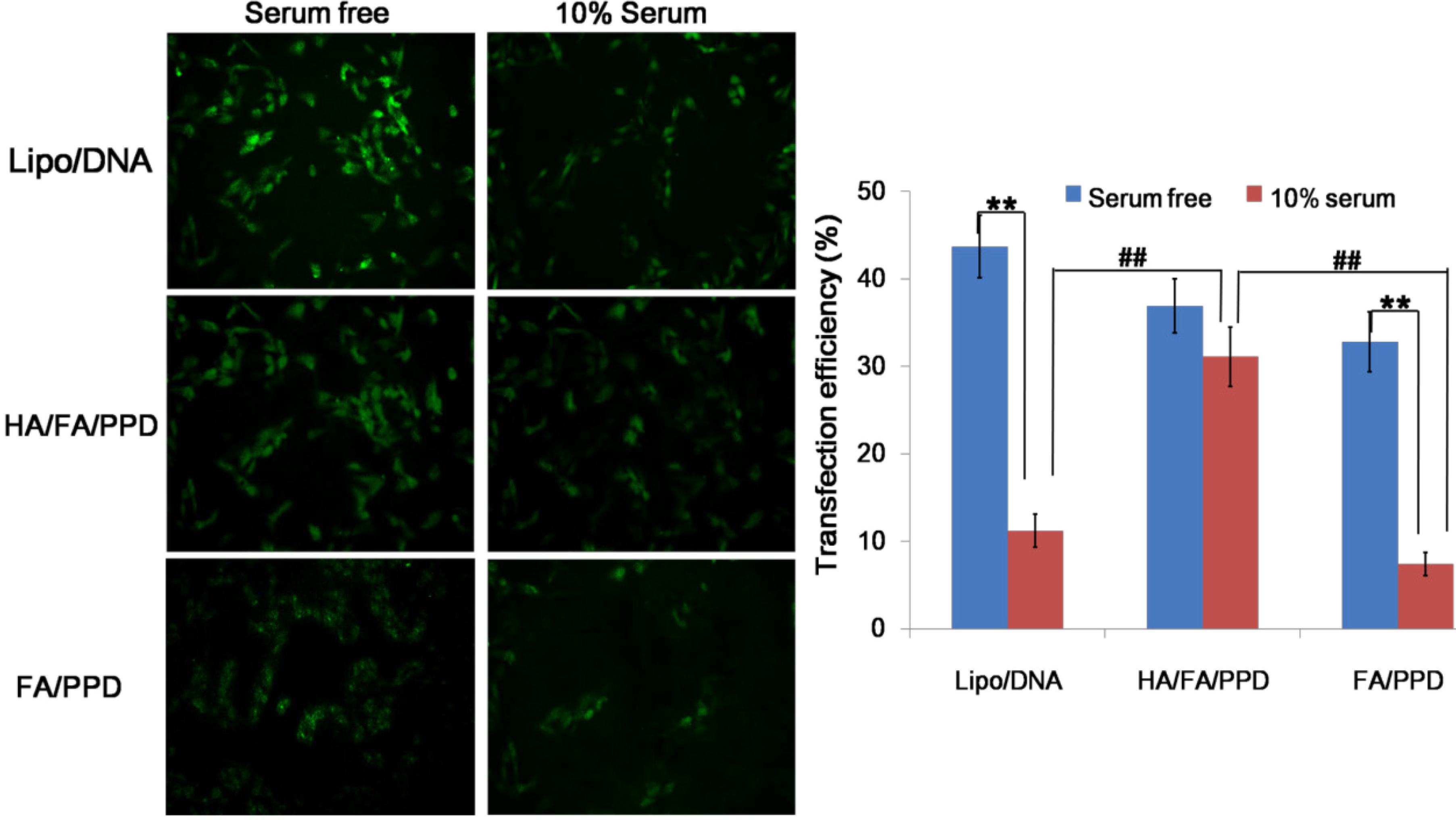
The transfection activity of HA/FA/PPD and FA/PPD on B16 cells was evaluated using plasmid pCMV-EGFP (pEGFP-N1) carrying enhanced green fluorescent protein (EGFP) as reporter gene. Briefly, B16 cells were seeded in six-well culture plates at a density of 3 × 105 cells per well and incubated overnight at 37 °C, 5% CO2. Before experiments, the growth media was removed and the cells were washed twice with pH 7.4, 0.01 M phosphate-buffered saline (PBS). Serum free cell culture media or 10% serum cell culture media containing HA/FA/PPD or FA/PPD were subsequently added. HA/FA/PPD and FA/PPD under different culture media were added at 2 μg of pDNA per well, respectively. After incubation for 4 h at 37 °C, the transfection media containing tested HA/FA/PPD or FA/PPD were removed and washed twice with PBS. Fresh RPM-1640 culture media containing 10% serum was added to each well and the cells were incubated for 48 h at 37 °C. Lipofectamine™ 2000/pDNA complexes containing equivalent pDNA were served as control. Fluorescence in cells was observed using an inverted fluorescence microscope (Olympus Corporation, Tokyo, Japan). After that, all cells were harvested and washed in PBS for three times, and the fluorescence intensity was quantitatively determined by a FACSCalibur™ fow cytometer (BD Biosciences, San Jose, CA, USA) by counting 10,000 events. Only the viable cells were gated for fluorescence analysis.
3.13. In Vitro Targeting Cellular Uptake Studies
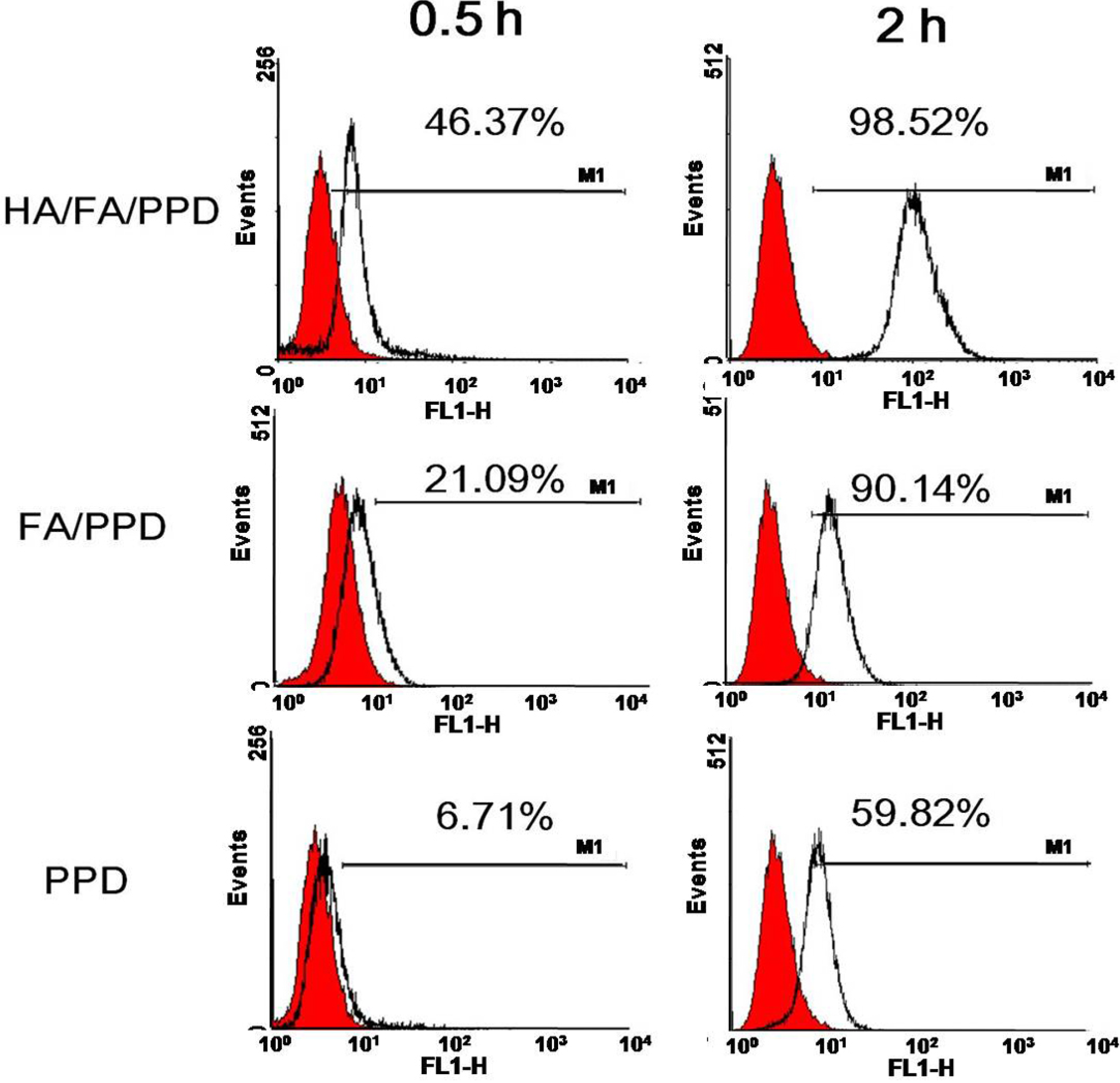
To investigate whether the dual targeting HA/FA/PPD showed advantages in cellular uptake, Rho-PTX labeled HA/FA/PPD, FA/PPD and PPD (10%,
w/
w) were prepared, respectively, and B16 cell was chosen as the CD44-positive and FA-positive cell line [
20,
21]. The cellular uptake of dual targeting HA/FA/PPD was quantified by flow cytometry. Briefly, B16 cells were seeded in 12-well culture plates at a density of 1 × 10
5 cells per well for overnight. After the cells reached 80% confluence, fresh media containing 2 μg/mL of Rho-PTX labeled HA/FA/PPD, FA/PPD and PPD were added, respectively, and incubated for 0.5 and 2 h at 37 °C, 5% CO
2 incubator. Then the media containing tested samples were removed and cells were harvested. The fluorescence intensity of the cells with different treatment was measured by flow cytometer (BD Biosciences). For each sample, 10,000 events were collected and the percentage of positive events was calculated as the events within the gate divided by the total number of events, excluding cell debris.
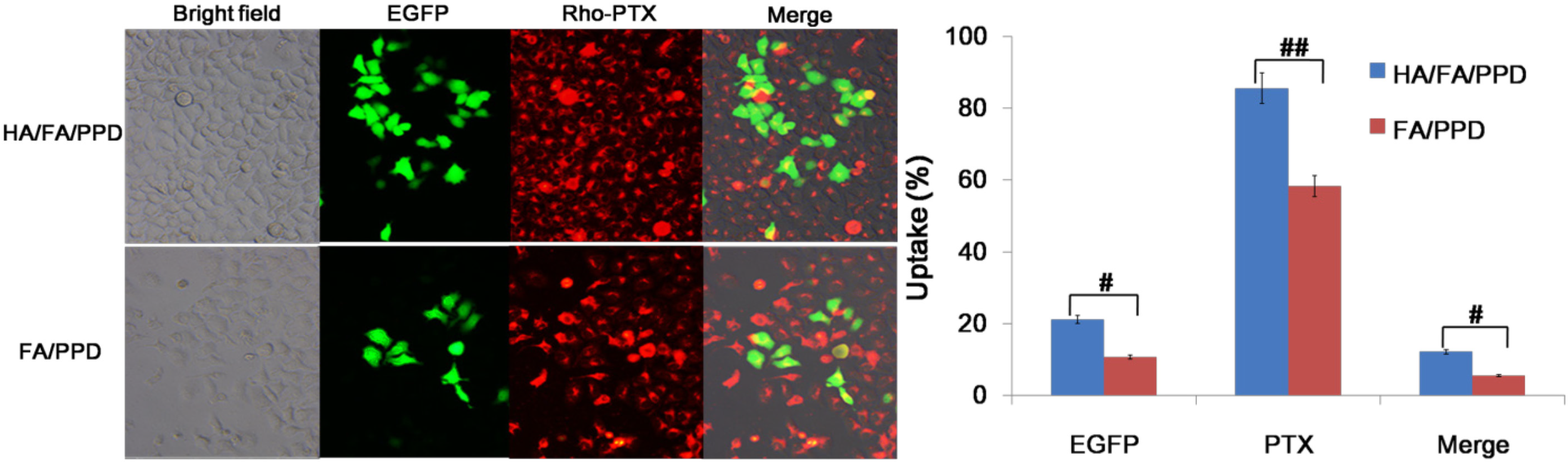
3.14. Co-Delivery of PTX and pDNA into Tumor Cells
To study whether pDNA and PTX could be co-delivered into the same cancer cell to achieve the potential synergistic cancer treatment, Rho-PTX labeled HA/FA/PPD, FA/PPD were prepared and HepG2 was chosen as the CD44-positive and FA-negative cell line [
22,
23]. Fluorescence microscope and a two-color flow cytometry were used to determine the co-delivery of PTX and pDNA. Briefly, HepG2 cells were seeded in six-well culture plates at a density of 3 × 10
5 cells per well and incubated overnight at 37 °C, 5% CO
2. Before experiments, the growth media was removed and the cells were washed twice with PBS. Serum free culture media containing HA/FA/PPD or FA/PPD were subsequently added. HA/FA/PPD and FA/PPD were added at 2 μg of pDNA and 2 μg/mL of Rho-PTX per well, respectively. After incubation for 4 h at 37 °C, the media containing tested HA/FA/PPD or FA/PPD were removed and washed twice with PBS buffer. Fresh RPM-1640 culture media containing 10% serum was added to each well and the cells were incubated for 48 h at 37 °C. Samples were observed under an inverted fluorescence microscope (Olympus Corporation, Tokyo, Japan) and quantitatively determined by a FACSCalibur™ flow cytometer (BD Biosciences).
3.15. Statistical Analysis
All studies were repeated a minimum of three times and measured at least in triplicate. Results were reported as mean ± SD (standard deviation). Statistical significance was analyzed using Student’s t-test. Differences between experimental groups were considered significant when p < 0.05.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









