
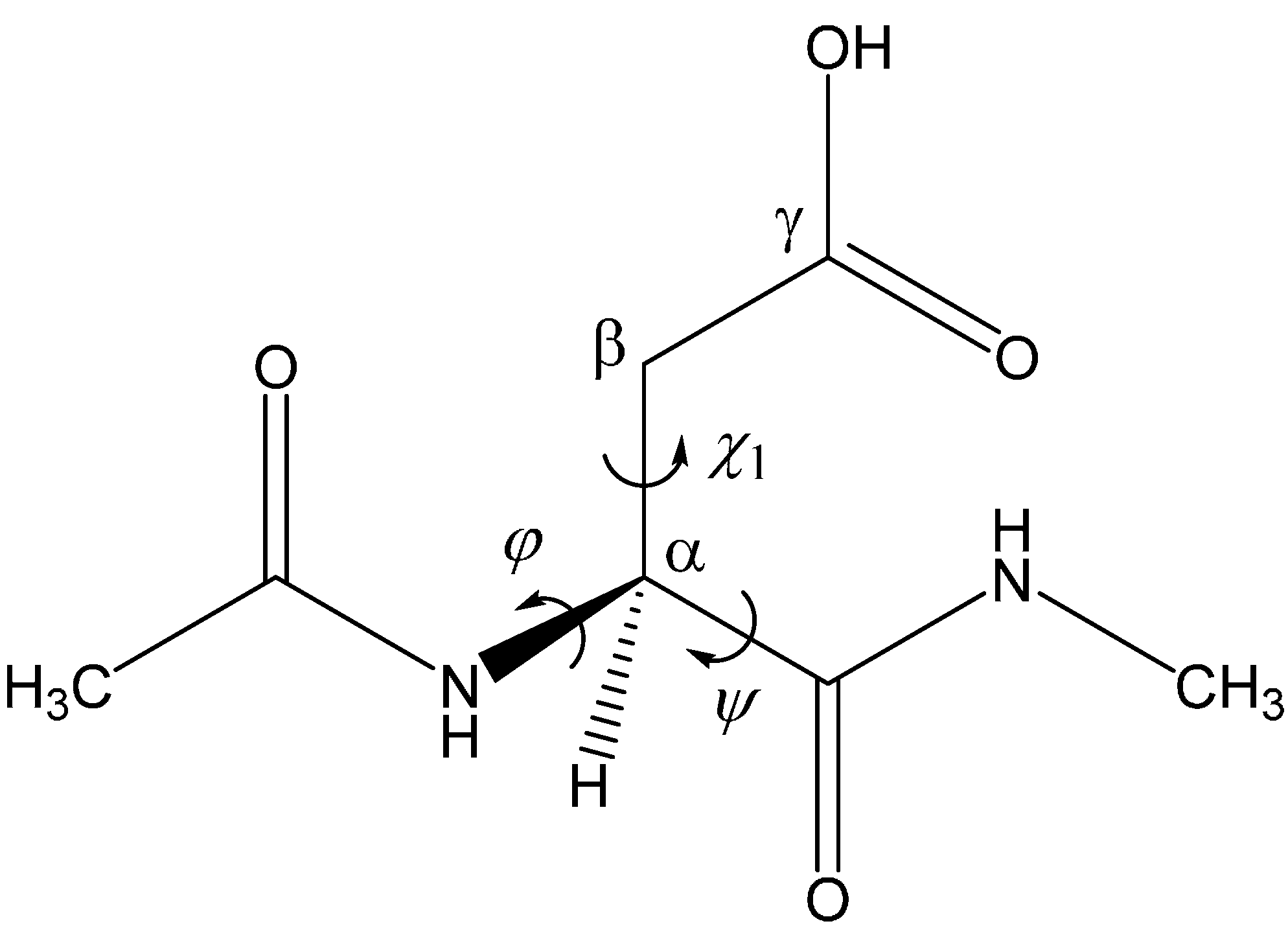
Acetic Acid Can Catalyze Succinimide Formation from Aspartic Acid Residues by a Concerted Bond Reorganization Mechanism: A Computational Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
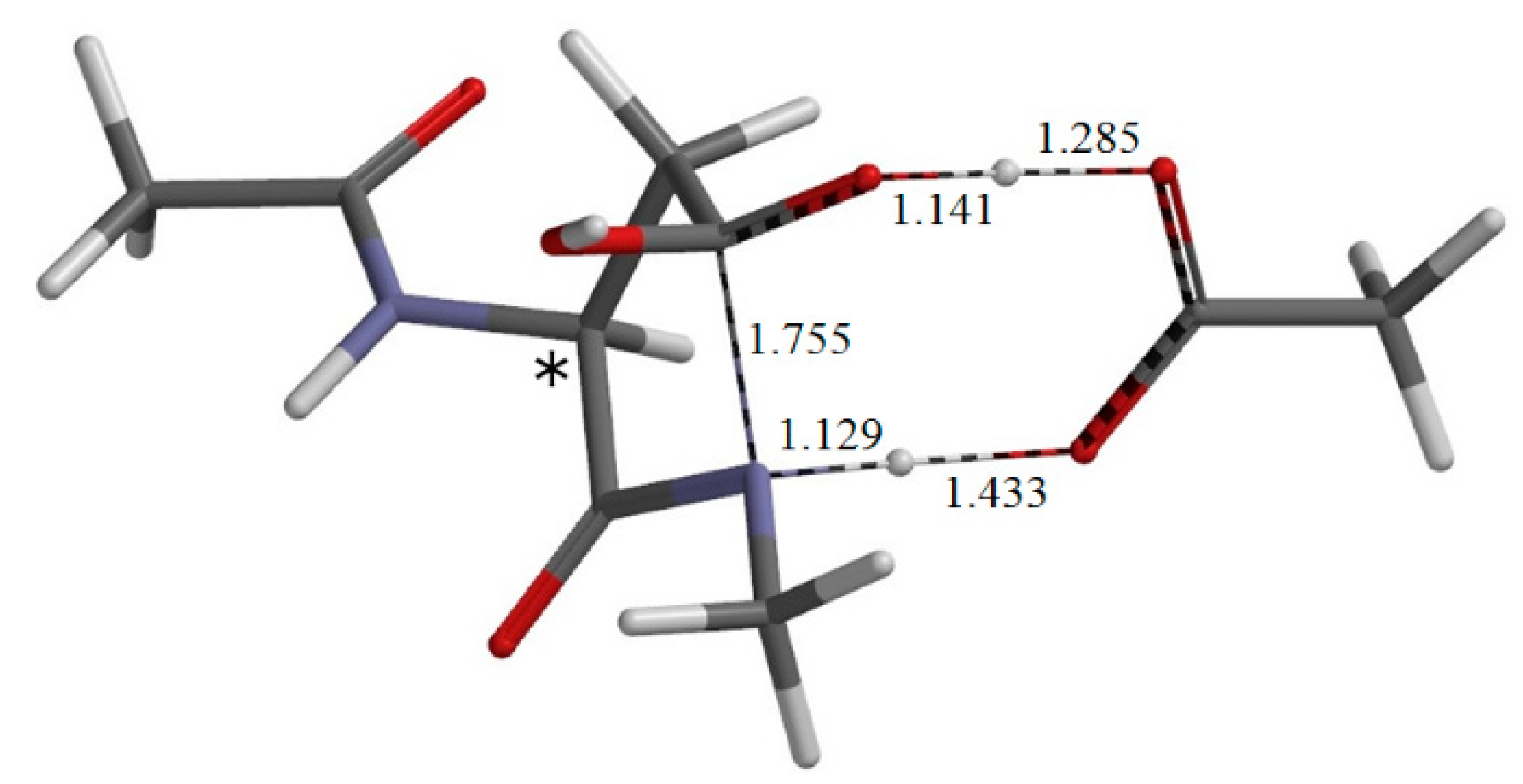{kind=link}
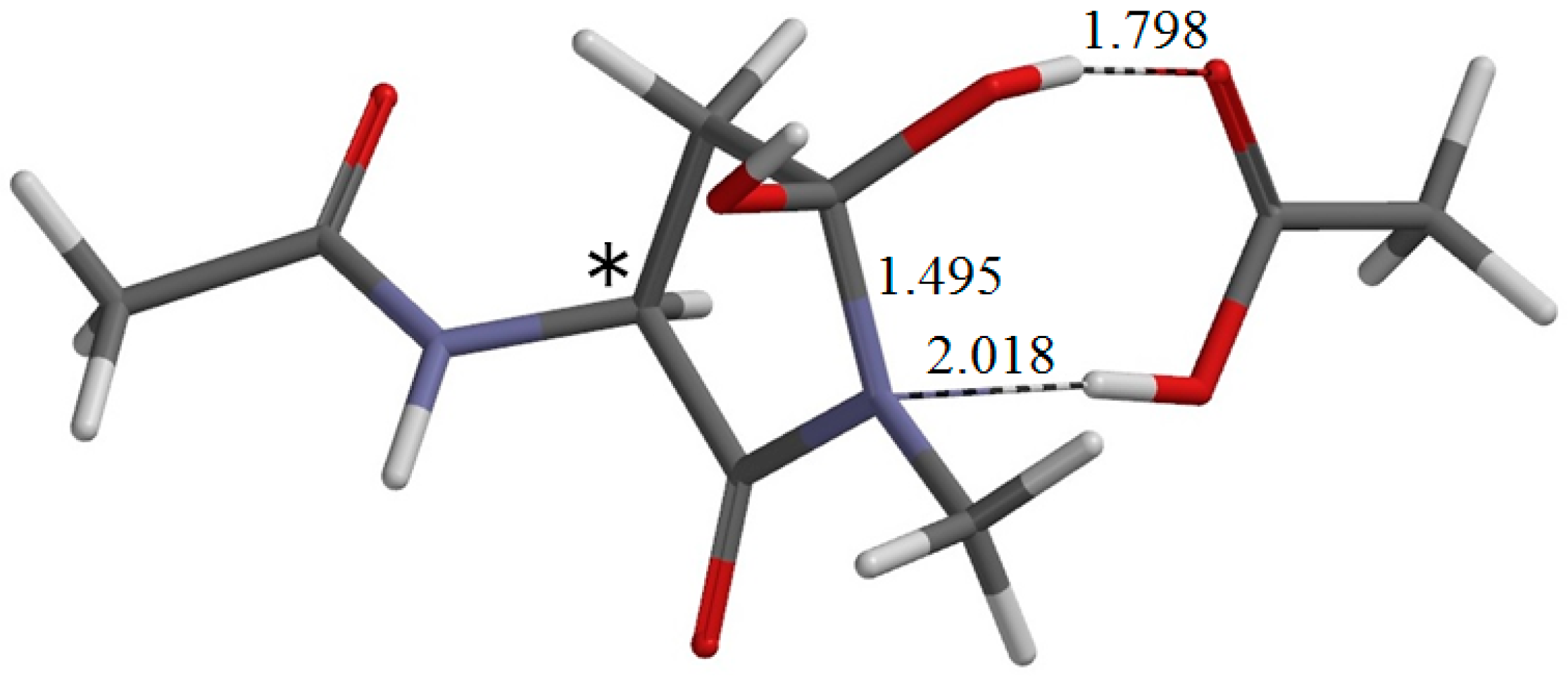{kind=link}
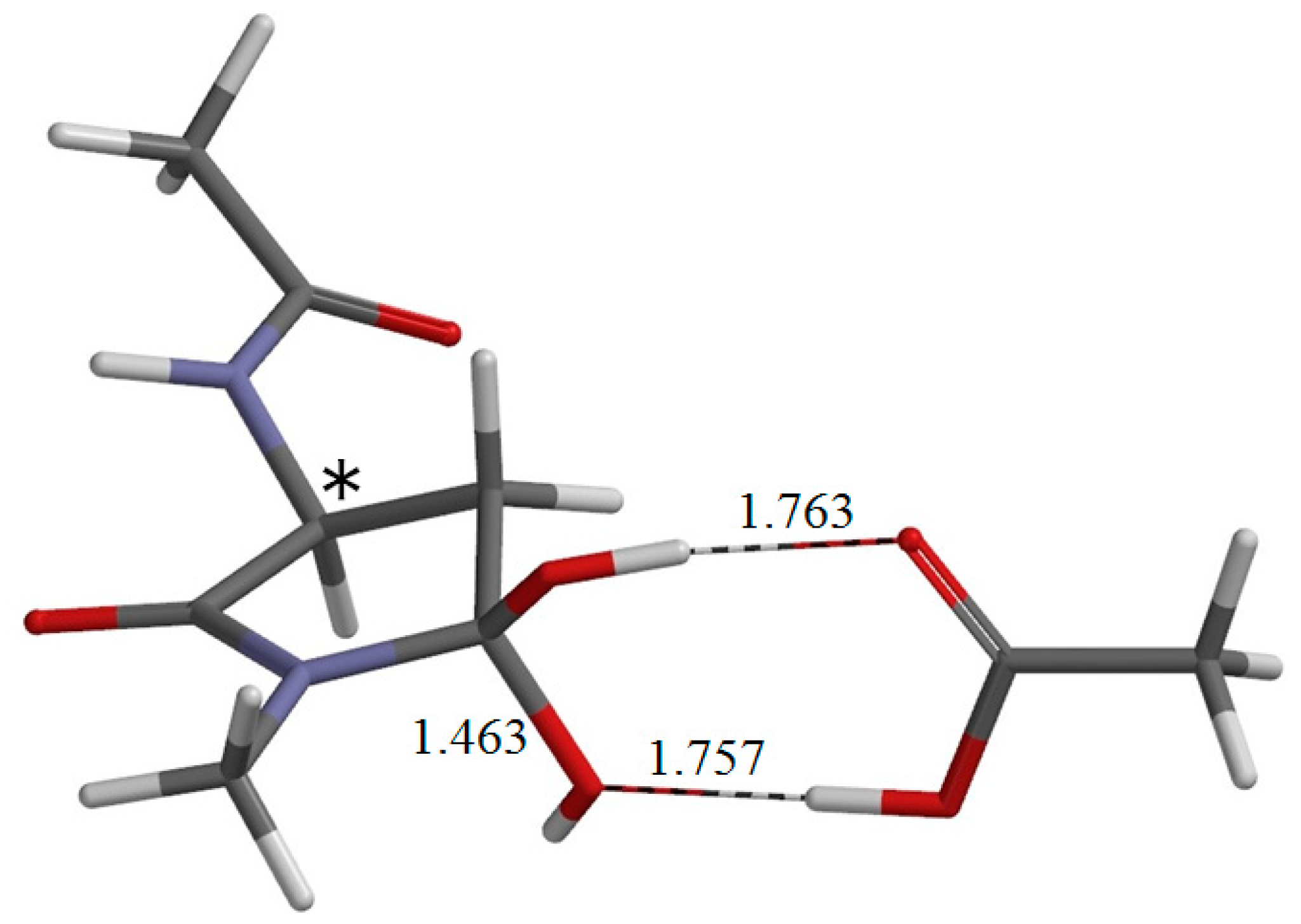{kind=link}
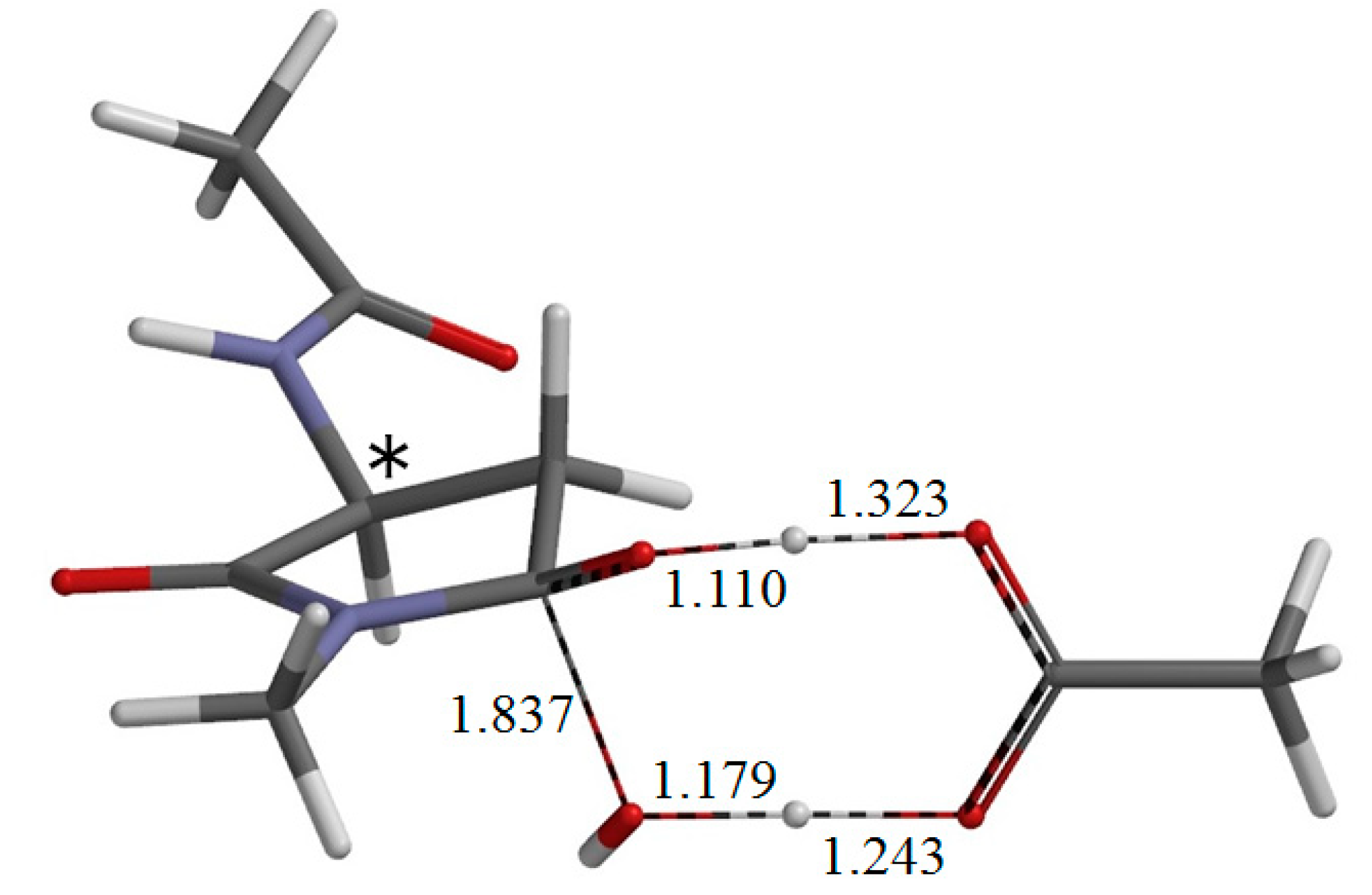{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
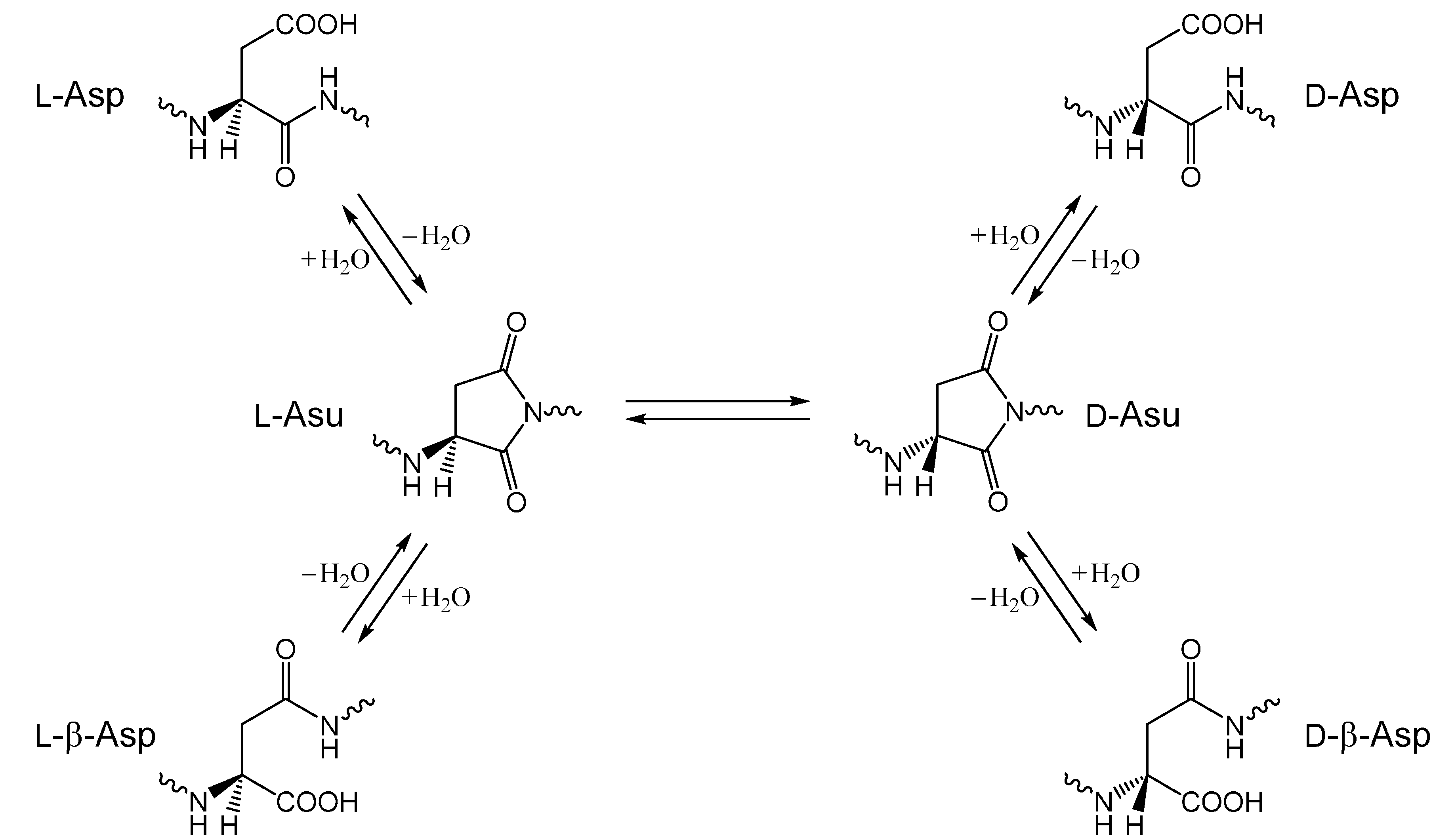
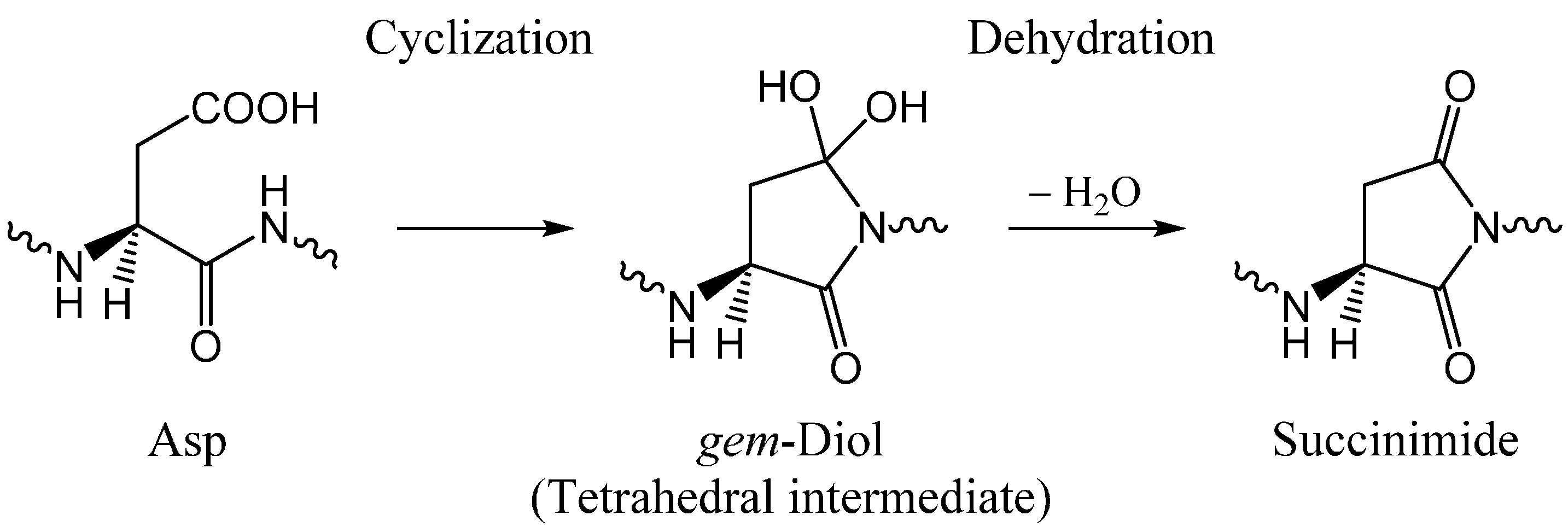
:1. Introduction



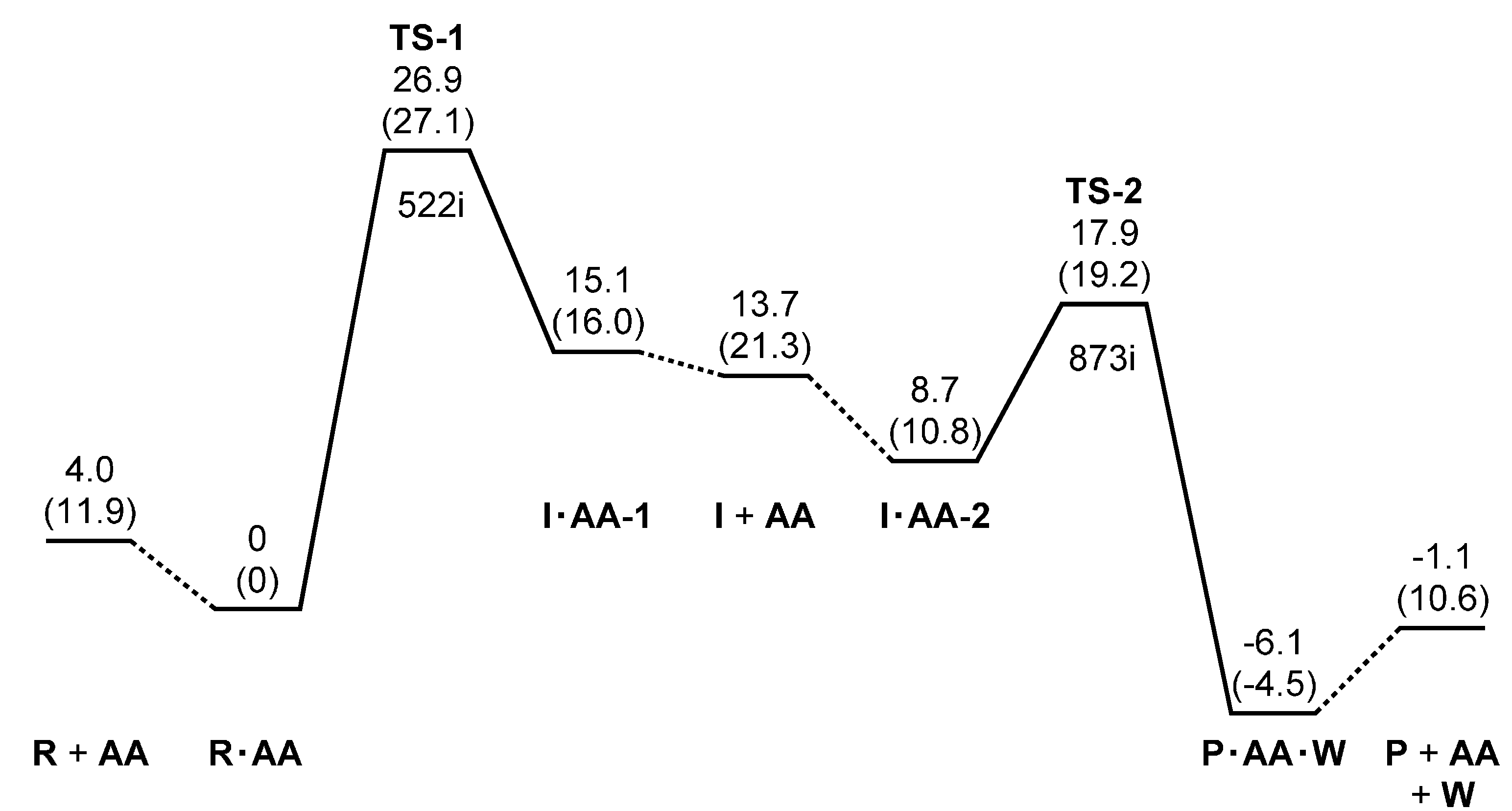
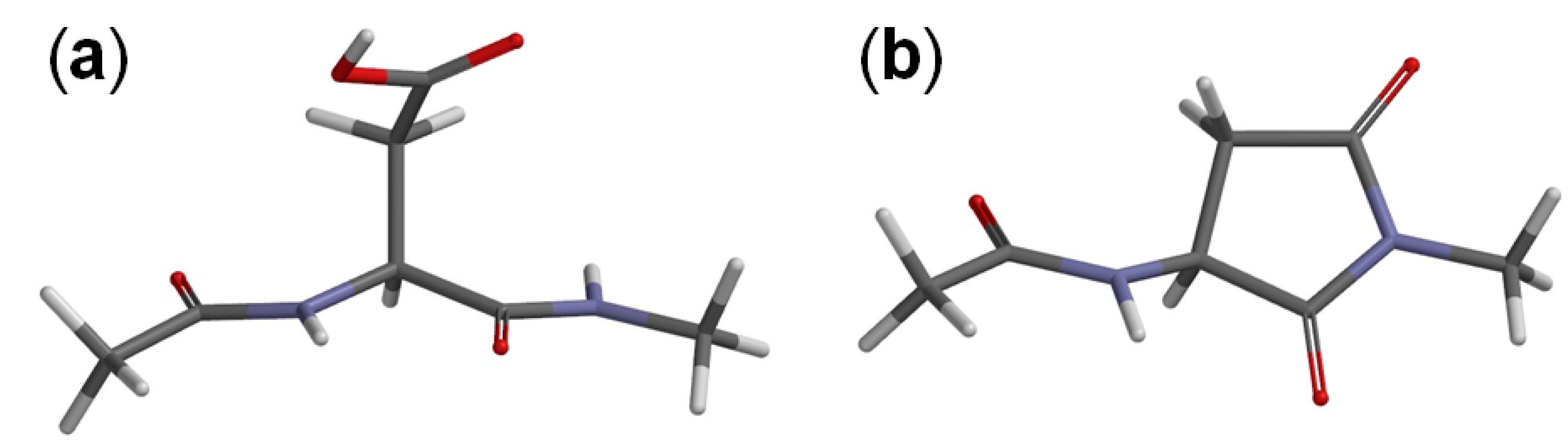
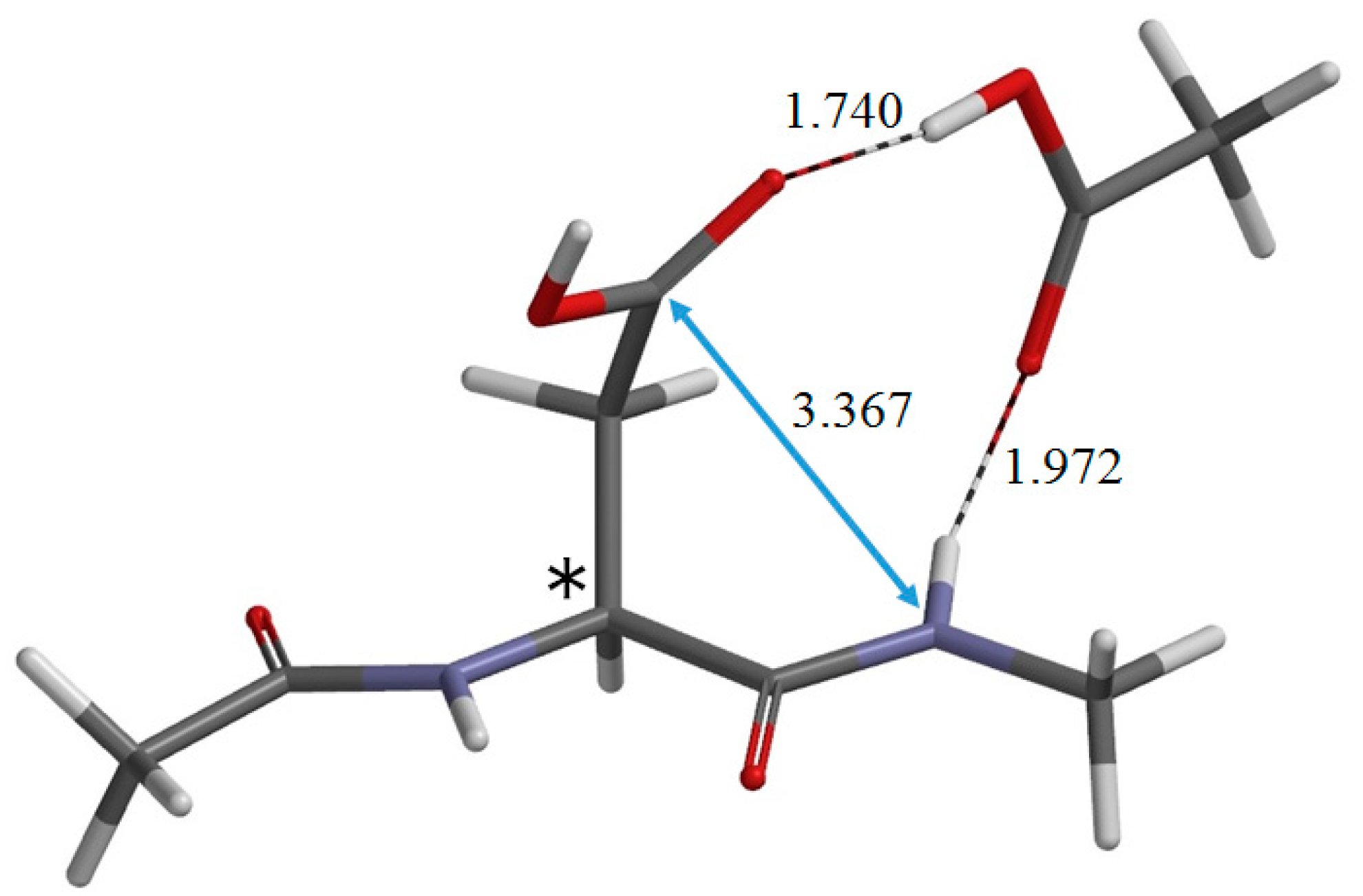
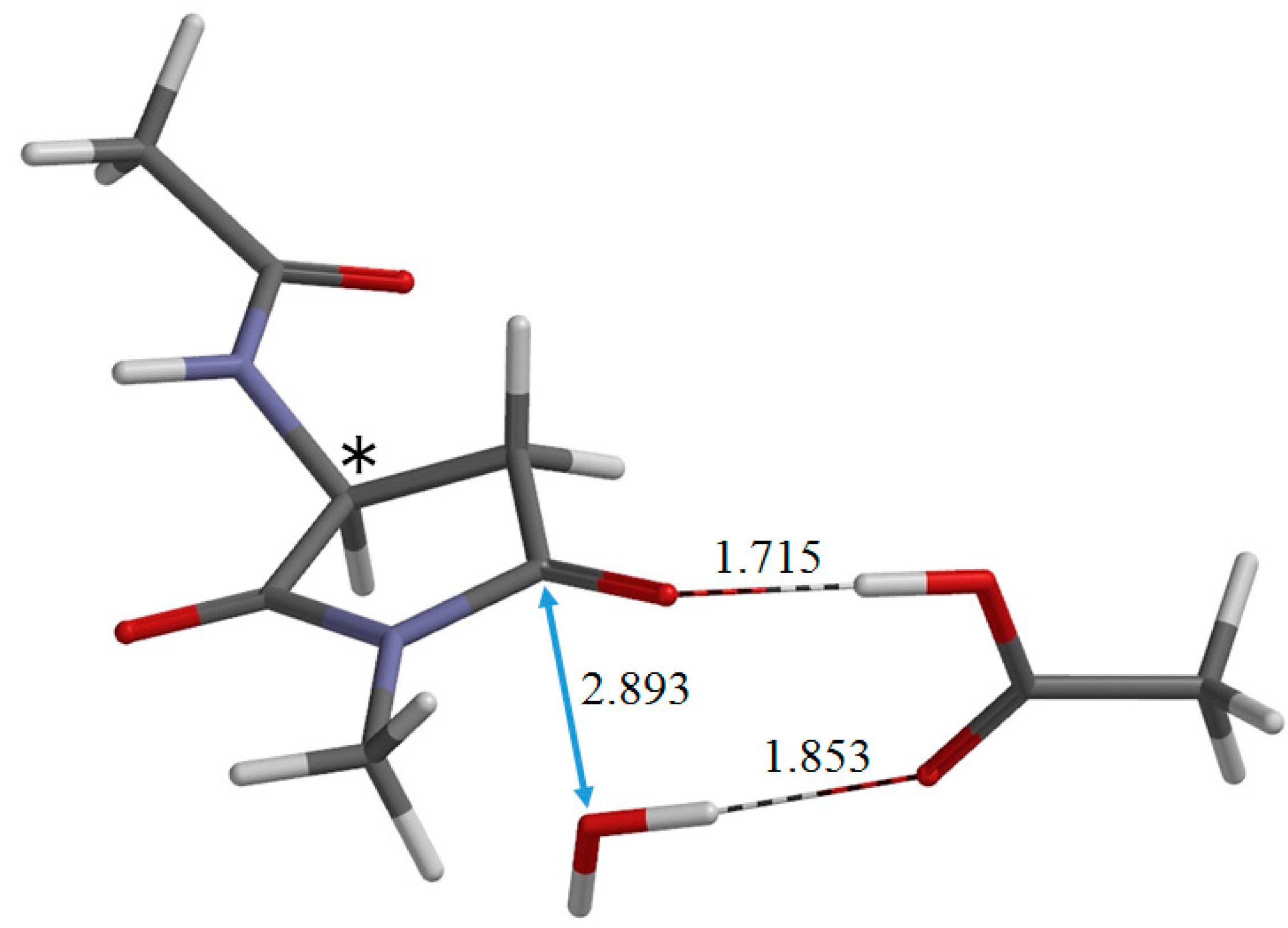
2. Results and Discussion








3. Computational Details
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Ritz-Timme, S.; Collins, M.J. Racemization of aspartic acid in human proteins. Aging Res. Rev. 2002, 1, 43–59. [Google Scholar] [CrossRef]
- Clarke, S. Aging as war between chemical and biochemical processes: Protein methylation and the recognition of age-damaged proteins for repair. Aging Res. Rev. 2003, 2, 263–285. [Google Scholar] [CrossRef]
- Fujii, N. d-Amino acid in elderly tissues. Biol. Pharm. Bull. 2005, 28, 1585–1589. [Google Scholar] [CrossRef] [PubMed]
- Truscott, R.J.W. Are ancient proteins responsible for the age-related decline in health and fitness? Rejuvenation Res. 2010, 13, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Takemoto, L.J.; Momose, Y.; Matsumoto, S.; Hiroki, K.; Akaboshi, M. Formation of four isomers at the Asp-151 residue of aged human αA-crystallin by natural aging. Biochem. Biophys. Res. Commun. 1999, 265, 746–751. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Matsumoto, S.; Hiroki, K.; Takemoto, L. Inversion and isomerization of Asp-58 residue in human αA-crystallin from normal aged lenses and cataractous lenses. Biochim. Biophys. Acta 2001, 1549, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Kawaguchi, T.; Sasaki, H.; Fujii, N. Simultaneous stereoinversion and isomerization at the Asp-4 residue in βB2-crystallin from the aged human eye lenses. Biochemistry 2011, 50, 8628–8635. [Google Scholar] [CrossRef] [PubMed]
- Hooi, M.Y.S.; Truscott, R.J.W. Racemisation and human cataract: d-Ser, d-Asp/Asn and d-Thr are higher in the lifelong proteins of cataract lenses than in age-matched normal lenses. Age 2011, 33, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Wolkow, C.; Wang, R.; Cotter, R.J.; Reardon, I.M.; Zürcher-Neely, H.A.; Heinrikson, R.L.; Ball, M.J.; et al. Structural alterations in the peptide backbone of β-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J. Biol. Chem. 1993, 268, 3072–3083. [Google Scholar] [PubMed]
- Tomiyama, T.; Asano, S.; Furiya, Y.; Shirasawa, T.; Endo, N.; Mori, H. Racemization of Asp23 residue affects the aggregation properties of Alzheimer amyloid β protein analogues. J. Biol. Chem. 1994, 269, 10205–10208. [Google Scholar] [PubMed]
- Sadakane, Y.; Konoha, K.; Kawahara, M.; Nakagomi, K. Quantification of structural alterations of l-Asp and l-Asn residues in peptides related to neuronal diseases by reversed-phase high-performance liquid chromatography. Chem. Biodivers. 2010, 7, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Galletti, P.; de Bonis, M.L.; Sorrentino, A.; Raimo, M.; D’Angelo, S.; Scala, I.; Andria, G.; D’Aniello, A.; Ingrosso, D.; Zappia, V. Accumulation of altered aspartyl residues in erythrocyte proteins from patients with Down’s syndrome. FEBS J. 2007, 274, 5263–5277. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.; Clarke, S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides: Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 1987, 262, 785–794. [Google Scholar] [PubMed]
- Stephenson, R.C.; Clarke, S. Succinimide formation from aspartyl and asparaginyl peptides as a model for the spontaneous degradation of proteins. J. Biol. Chem. 1989, 264, 6164–6170. [Google Scholar] [PubMed]
- Radkiewicz, J.L.; Zipse, H.; Clarke, S.; Houk, K.N. Accelerated racemization of aspartic acid and asparagine residues via succinimide intermediates: An ab initio theoretical exploration of mechanism. J. Am. Chem. Soc. 1996, 118, 9148–9155. [Google Scholar] [CrossRef]
- Takahashi, O.; Kobayashi, K.; Oda, A. Modeling the enolization of succinimide derivatives, a key step of racemization of aspartic acid residues: Importance of a two-H2O mechanism. Chem. Biodivers. 2010, 7, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, O. Two-water-assisted racemization of the succinimide intermediate formed in proteins: A computational model study. Health 2013, 5, 2018–2021. [Google Scholar] [CrossRef]
- Capasso, S. Thermodynamic parameters of the reversible isomerization of aspartic residues via a succinimide derivative. Thermochim. Acta 1996, 286, 41–50. [Google Scholar] [CrossRef]
- Reissner, K.J.; Aswad, D.W. Deamidation and isoaspartate formation in proteins: Unwanted alterations or surreptitious signals? Cell. Mol. Life Sci. 2003, 60, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Aylin, F.; Konuklar, S.; Aviyente, V. Modelling the hydrolysis of succinimide: Formation of aspartate and reversible isomerization of aspartic acid via succinimide. Org. Biomol. Chem. 2003, 1, 2290–2297. [Google Scholar] [CrossRef] [PubMed]
- Catak, S.; Monard, G.; Aviyente, V.; Ruiz-López, M.F. Deamidation of asparagine residues: Direct hydrolysis versus succinimide-mediated deamidation mechanisms. J. Phys. Chem. A 2009, 113, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, O.; Oda, A. Amide-iminol tautomerization of the C-terminal peptide groups of aspartic acid residues: Two-water-assisted mechanism, cyclization from the iminol tautomer leading to the tetrahedral intermediate of succinimide formation, and implication to peptide group hydrogen exchange. In Tyrosine and Aspartic Acid: Properties, Sources and Health Benefits; Jones, J.E., Morano, D.M., Eds.; Nova Science Publishers: New York, NY, USA, 2012; pp. 131–147. [Google Scholar]
- Takahashi, O.; Kirikoshi, R. Intramolecular cyclization of aspartic acid residues assisted by three water molecules: A density functional theory study. Comput. Sci. Discov. 2014, 7, 015005. [Google Scholar] [CrossRef]
- Takahashi, O.; Kirikoshi, R.; Manabe, N. Roles of intramolecular and intermolecular hydrogen bonding in a three-water-assisted mechanism of succinimide formation from aspartic acid residues. Molecules 2014, 19, 11440–11452. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, H.; Yamada, H.; Ueda, T.; Imoto, T. Isolation and characterization of 101-succinimide lysozyme that possesses the cyclic imide at Asp101-Gly102. Biochemistry 1994, 33, 8770–8774. [Google Scholar] [CrossRef] [PubMed]
- Vaney, M.C.; Maignan, S.; Riès-Kautt, M.; Ducruix, A. High-resolution structure (1.33 Å) of a HEW lysozyme tetragonal crystal grown in the APCF apparatus: Data and structural comparison with a crystal grown under microgravity from SpaceHab-01 mission. Acta Crystallogr. D Biol. Crystallogr. 1996, 52, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, K.; Noguchi, S.; Harada, S.; Satow, Y. Crystallography of succinimide hen egg-white lysozyme at low temperatures. J. Cryst. Growth 1996, 168, 292–296. [Google Scholar] [CrossRef]
- Noguchi, S.; Miyawaki, K.; Satow, Y. Succinimide and isoaspartate residues in the crystal structures of hen egg-white lysozyme complexed with tri-N-acetylchitotriose. J. Mol. Biol. 1998, 278, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, A. Understanding Organic Reaction Mechanisms; Cambridge University Press: Cambridge, UK, 1997; p. 131. [Google Scholar]
- Perry, C.J.; Parveen, Z. The cyclisation of substituted phthalanilic acids in acetic acid solution: A kinetic study of substituted N-phenylphthalimide formation. J. Chem. Soc. Perkin Trans. 2 2001, 512–521. [Google Scholar] [CrossRef]
- Tanaka, H.; Iwata, Y.; Takahashi, D.; Adachi, M.; Takahashi, T. Efficient stereoselective synthesis of γ-N-glycosyl asparagines by N-glycosylation of primary amide groups. J. Am. Chem. Soc. 2005, 127, 1630–1631. [Google Scholar] [CrossRef] [PubMed]
- Bacchi, A.; Costa, M.; Della Cà, N.; Gabriele, B.; Salerno, G.; Cassoni, S. Heterocyclic derivative syntheses by palladium-catalyzed oxidative cyclization-alkoxycarbonylation of substituted γ-oxoalkynes. J. Org. Chem. 2005, 70, 4971–4979. [Google Scholar] [CrossRef] [PubMed]
- Pathak, S.; Kundu, A.; Pramanik, A. Regioselective synthesis of two types of highly substituted 2-pyridones through similar multicomponent reactions. Tetrahedron Lett. 2012, 53, 3030–3034. [Google Scholar] [CrossRef]
- Panda, N.; Mothkuri, R. Stereoselective synthesis of enamides by Pd-catalyzed hydroamidation of electron deficient terminal alkynes. J. Org. Chem. 2012, 77, 9407–9412. [Google Scholar] [CrossRef] [PubMed]
- Powell, M.F. A compendium and hydropathy/flexibility analysis of common reactive sites in proteins: Reactivity at Asn, Asp, Gln, and Met motifs in neutral pH solution. In Formulation, Characterization, and Stability of Protein Drugs; Pearlman, R., Wang, Y.J., Eds.; Springer Science + Business Media: New York, NY, USA, 1996; pp. 1–140. [Google Scholar]
- Cacia, J.; Keck, R.; Presta, L.G.; Frenz, J. Isomerization of an aspartic acid residue in the complementarity-determining regions of a recombinant antibody to human IgE: Identification and effect on binding affinity. Biochemistry 1996, 35, 1897–1903. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Instability, stabilization, and formulation of liquid protein pharmaceuticals. Int. J. Pharm. 1999, 185, 129–188. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.J.; Kabakoff, B.; Macchi, F.D.; Shen, F.J.; Kwong, M.; Andya, J.D.; Shire, S.J.; Bjork, N.; Totpal, K.; Chen, A.B. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J. Chromatogr. B 2001, 752, 233–245. [Google Scholar] [CrossRef]
- Wakankar, A.A.; Borchardt, R.T. Formulation considerations for proteins susceptible to asparagine deamidation and aspartate isomerization. J. Pharm. Sci. 2006, 95, 2321–2336. [Google Scholar] [CrossRef] [PubMed]
- Wakankar, A.A.; Borchardt, R.T.; Eigenbrot, C.; Shia, S.; Wang, Y.J.; Shire, S.J.; Liu, J.L. Aspartate isomerization in the complementarity-determining regions of two closely related monoclonal antibodies. Biochemistry 2007, 46, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Bondarenko, P.V.; Jacob, J.; Chu, G.C.; Chelius, D. 18O labeling method for identification and quantification of succinimide in proteins. Anal. Chem. 2007, 79, 2714–2721. [Google Scholar] [CrossRef] [PubMed]
- Chu, G.C.; Chelius, D.; Xiao, G.; Khor, H.K.; Coulibaly, S.; Bondarenko, P.V. Accumulation of succinimide in a recombinant monoclonal antibody in mildly acidic buffers under elevated temperatures. Pharm. Res. 2007, 24, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Bondarenko, P.V. Identification and quantification of degradations in the Asp-Asp motifs of a recombinant monoclonal antibody. J. Pharm. Biomed. Anal. 2008, 47, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Valliere-Douglass, J.; Jones, L.; Shpektor, D.; Kodama, P.; Wallace, A.; Balland, A.; Bailey, R.; Zhang, Y. Separation and characterization of an IgG2 antibody containing a cyclic imide in CDR1 of light chain by hydrophobic interaction chromatography and mass spectrometry. Anal. Chem. 2008, 80, 3168–3174. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Z.; Nichols, A.; Liu, D. Direct identification and quantification of aspartyl succinimide in an IgG2 mAb by RapiGest assisted digestion. Anal. Chem. 2009, 81, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yip, H.; Katta, V. Identification of isomerization and racemization of aspartate in the Asp-Asp motifs of a therapeutic protein. Anal. Biochem. 2011, 410, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.C.; Joe, K.; Zhang, Y.; Adriano, A.; Wang, Y.; Gazzano-Santoro, H.; Keck, R.G.; Deperalta, G.; Ling, V. Accurate determination of succinimide degradation products using high fidelity trypsin digestion peptide map analysis. Anal. Chem. 2011, 83, 5912–5919. [Google Scholar] [CrossRef] [PubMed]
- Sreedhara, A.; Cordoba, A.; Zhu, Q.; Kwong, J.; Liu, J. Characterization of the isomerization products of aspartate residues at two different sites in a monoclonal antibody. Pharm. Res. 2012, 29, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Beckley, N.; Gikanga, B.; Zhang, J.; Wang, Y.J.; Chih, H.W.; Sharma, V.K. Isomerization of Asp-Asp motif in model peptides and a monoclonal antibody Fab fragment. J. Pharm. Sci. 2013, 102, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Herman, A.C.; Boone, T.C.; Lu, H.S. Characterization, formulation, and stability of Neupogen® (Filgrastim), a recombinant human granulocyte-colony stimulating factor. In Formulation, Characterization, and Stability of Protein Drugs; Pearlman, R., Wang, Y.J., Eds.; Springer Science + Business Media: New York, NY, USA, 1996; pp. 303–328. [Google Scholar]
- Lam, X.M.; Yang, J.Y.; Cleland, J.L. Antioxidants for prevention of methionine oxidation in recombinant monoclonal antibody HER2. J. Pharm. Sci. 1997, 86, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- Shahrokh, Z.; Eberlein, G.; Buckley, D.; Paranandi, M.V.; Aswad, D.W.; Stratton, P.; Mischak, R.; Wang, Y.J. Major degradation products of basic fibroblast growth factor: Detection of succinimide and iso-aspartate in place of aspartate15. Pharm. Res. 1994, 11, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Vander Velde, D.; Morton, M.; Borchardt, R.T.; Schowen, R.L. pH-Induced change in the rate-determining step for the hydrolysis of the Asp/Asn-derived cyclic-imide intermediate in protein degradation. J. Am. Chem. Soc. 1996, 118, 8955–8956. [Google Scholar] [CrossRef]
- Violand, B.N.; Schlittler, M.R.; Kolodziej, E.W.; Toren, P.C.; Cabonce, M.A.; Siegel, N.R.; Duffin, K.L.; Zobel, J.F.; Smith, C.E.; Tou, J.S. Isolation and characterization of porcine somatotropin containing a succinimide residue in place of aspartate129. Protein Sci. 1992, 1, 1634–1641. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Frey, W.; Peters, R. Asymmetric palladium(II)-catalyzed cascade reaction giving quaternary amino succinimides by 1,4-addition and a Nef-type reaction. Angew. Chem. Int. Ed. 2013, 52, 13223–13227. [Google Scholar] [CrossRef]
- Marenich, A.V.; Olson, R.M.; Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. Self-consistent reaction field model for aqueous and nonaqueous solutions based on accurate polarized partial charges. J. Chem. Theory Comput. 2007, 3, 2011–2033. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G. A universal approach to solvation modeling. Acc. Chem. Res. 2008, 41, 760–768. [Google Scholar] [CrossRef]
- Ben-Naim, A.; Marcus, Y. Solvation thermodynamics of nonionic solutes. J. Chem. Phys. 1984, 81, 2016–2027. [Google Scholar] [CrossRef]
- Kim, Y. Direct dynamics calculation for the double proton transfer in formic acid dimer. J. Am. Chem. Soc. 1996, 118, 1522–1528. [Google Scholar] [CrossRef]
- Loerting, T.; Liedl, K.R. Toward elimination of discrepancies between theory and experiment: Double proton transfer in dimers of carboxylic acids. J. Am. Chem. Soc. 1998, 120, 12595–12600. [Google Scholar] [CrossRef]
- Durlak, P.; Berski, S.; Latajka, Z. Car–Parrinello and path integral molecular dynamics study of the hydrogen bond in the acetic acid dimer in the gas phase. J. Mol. Model. 2011, 17, 2995–3004. [Google Scholar] [CrossRef] [PubMed]
- Chou, P.T.; Chen, Y.C.; Wei, C.Y.; Chen, W.S. Excited-state amino-imino double-proton tautomerism in adenine nucleotide analogues catalyzed by carboxylic acids. J. Am. Chem. Soc. 2000, 122, 9322–9323. [Google Scholar] [CrossRef]
- Hung, F.T.; Hu, W.P.; Chou, P.T. The ground- and excited-state (1nπ* and 1ππ*) carboxylic acid-catalyzed proton (hydrogen atom)-transfer energy surfaces in 3-formyl-7-azaindole. J. Phys. Chem. A 2001, 105, 10475–10482. [Google Scholar] [CrossRef]
- Sikorska, E.; Khmelinskii, I.; Hoffmann, M.; Machado, I.F.; Ferreira, L.F.V.; Dobek, K.; Karolczak, J.; Krawczyk, A.; Insińska-Rak, M.; Sikorski, M. Ground- and excited-state double proton transfer in lumichrome/acetic acid system: Theoretical and experimental approach. J. Phys. Chem. A 2005, 109, 11707–11714. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.; Zhao, G.J.; Song, P.; Yang, S.Q.; Liu, J.Y.; Han, K.L. Reconsideration of the excited-state double proton transfer (ESDPT) in 2-aminopyridine/acid systems: Role of the intermolecular hydrogen bonding in excited states. Phys. Chem. Chem. Phys. 2009, 11, 4385–4390. [Google Scholar] [CrossRef] [PubMed]
- Catak, S.; Monard, G.; Aviyente, V.; Ruiz-López, M.F. Reaction mechanism of deamidation of asparaginyl residues in peptides: Effect of solvent molecules. J. Phys. Chem. A 2006, 110, 8354–8365. [Google Scholar] [CrossRef] [PubMed]
- Spartan’14, version 1.1.4; Wavefunction, Inc.: Irvine, CA, USA, 2014.
- Connolly, B.D.; Tran, B.; Moore, J.M.R.; Sharma, V.K.; Kosky, A. Specific catalysis of asparaginyl deamidation by carboxylic acids: Kinetic, thermodynamic, and quantitative structure-property relationship analyses. Mol. Pharm. 2014, 11, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahashi, O.; Kirikoshi, R.; Manabe, N. Acetic Acid Can Catalyze Succinimide Formation from Aspartic Acid Residues by a Concerted Bond Reorganization Mechanism: A Computational Study. Int. J. Mol. Sci. 2015, 16, 1613-1626. https://doi.org/10.3390/ijms16011613
Takahashi O, Kirikoshi R, Manabe N. Acetic Acid Can Catalyze Succinimide Formation from Aspartic Acid Residues by a Concerted Bond Reorganization Mechanism: A Computational Study. International Journal of Molecular Sciences. 2015; 16(1):1613-1626. https://doi.org/10.3390/ijms16011613
Chicago/Turabian StyleTakahashi, Ohgi, Ryota Kirikoshi, and Noriyoshi Manabe. 2015. "Acetic Acid Can Catalyze Succinimide Formation from Aspartic Acid Residues by a Concerted Bond Reorganization Mechanism: A Computational Study" International Journal of Molecular Sciences 16, no. 1: 1613-1626. https://doi.org/10.3390/ijms16011613
APA StyleTakahashi, O., Kirikoshi, R., & Manabe, N. (2015). Acetic Acid Can Catalyze Succinimide Formation from Aspartic Acid Residues by a Concerted Bond Reorganization Mechanism: A Computational Study. International Journal of Molecular Sciences, 16(1), 1613-1626. https://doi.org/10.3390/ijms16011613