
Motor, Visual and Emotional Deficits in Mice after Closed-Head Mild Traumatic Brain Injury Are Alleviated by the Novel CB2 Inverse Agonist SMM-189

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Cultured Human Microglia
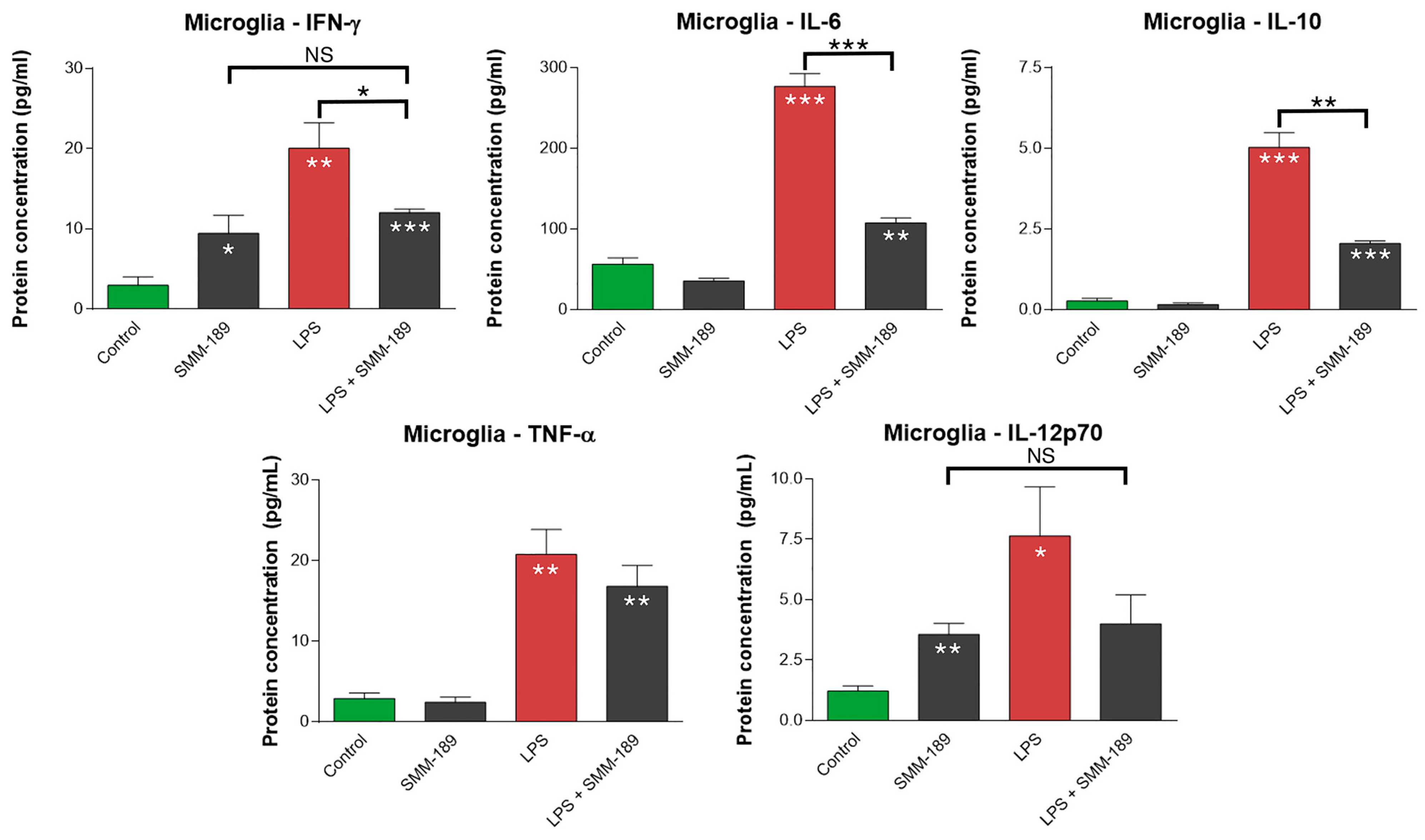
2.1.1. Cytokines

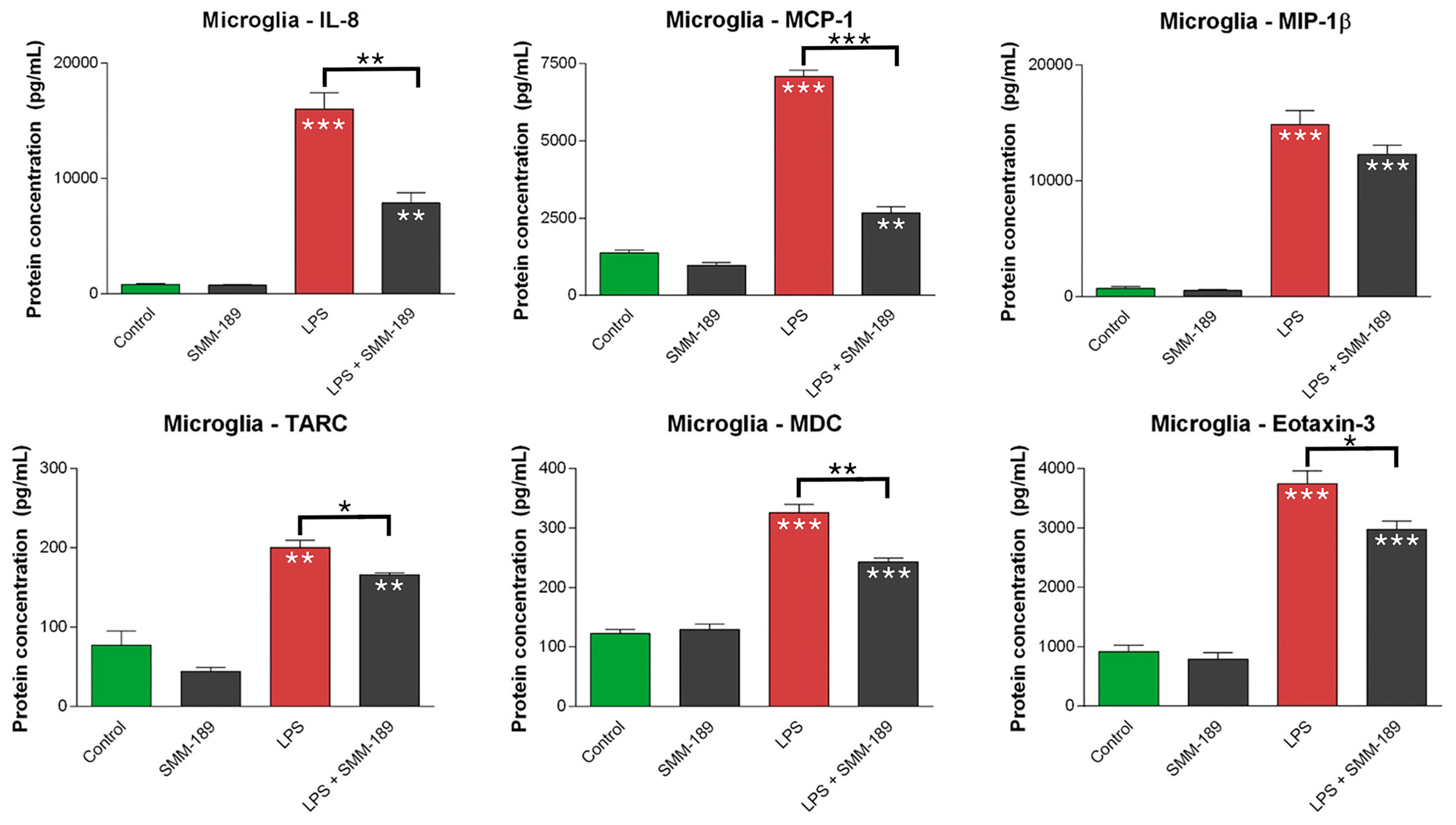
2.1.2. Chemokines

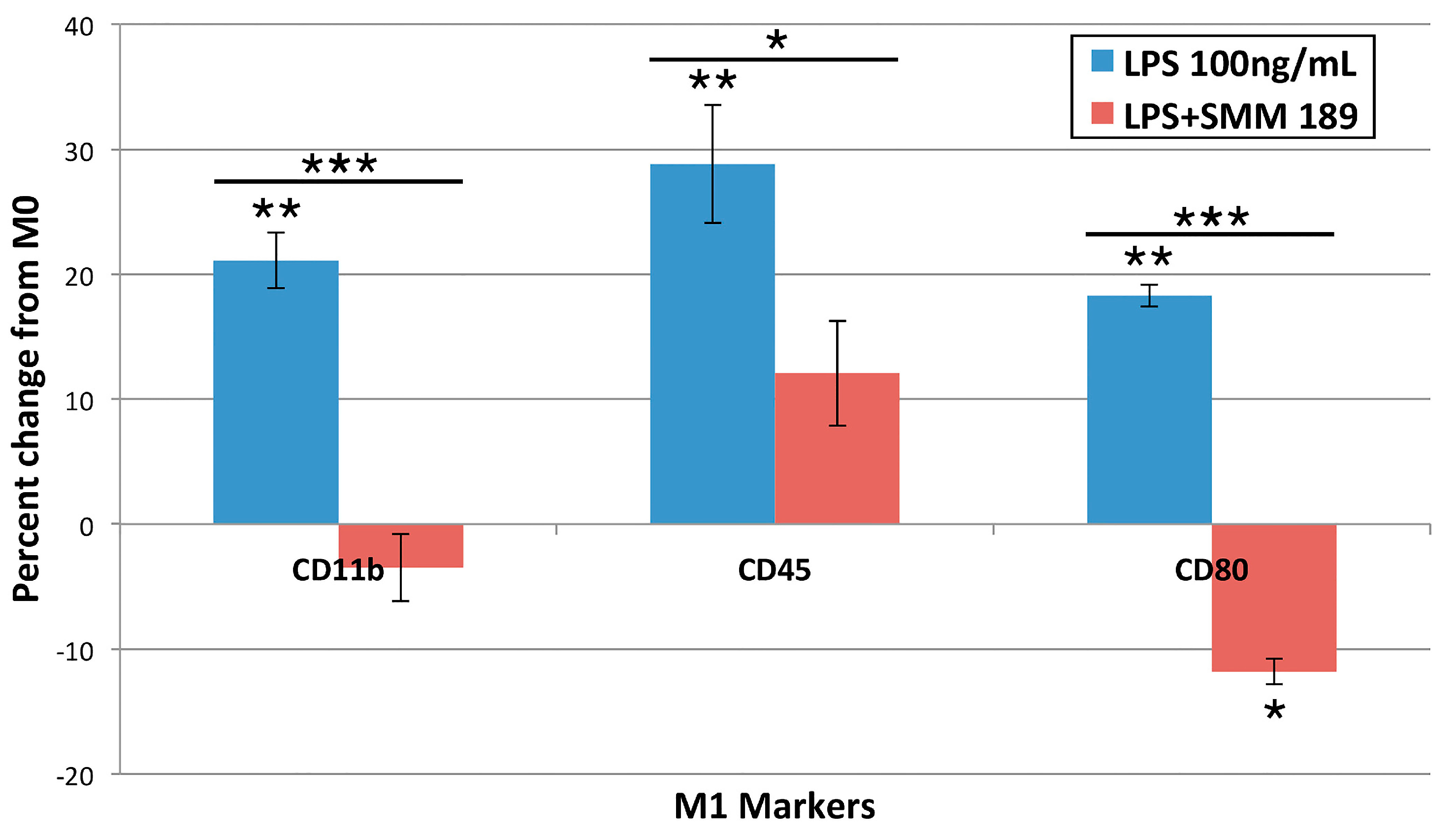
2.1.3. M1 Markers

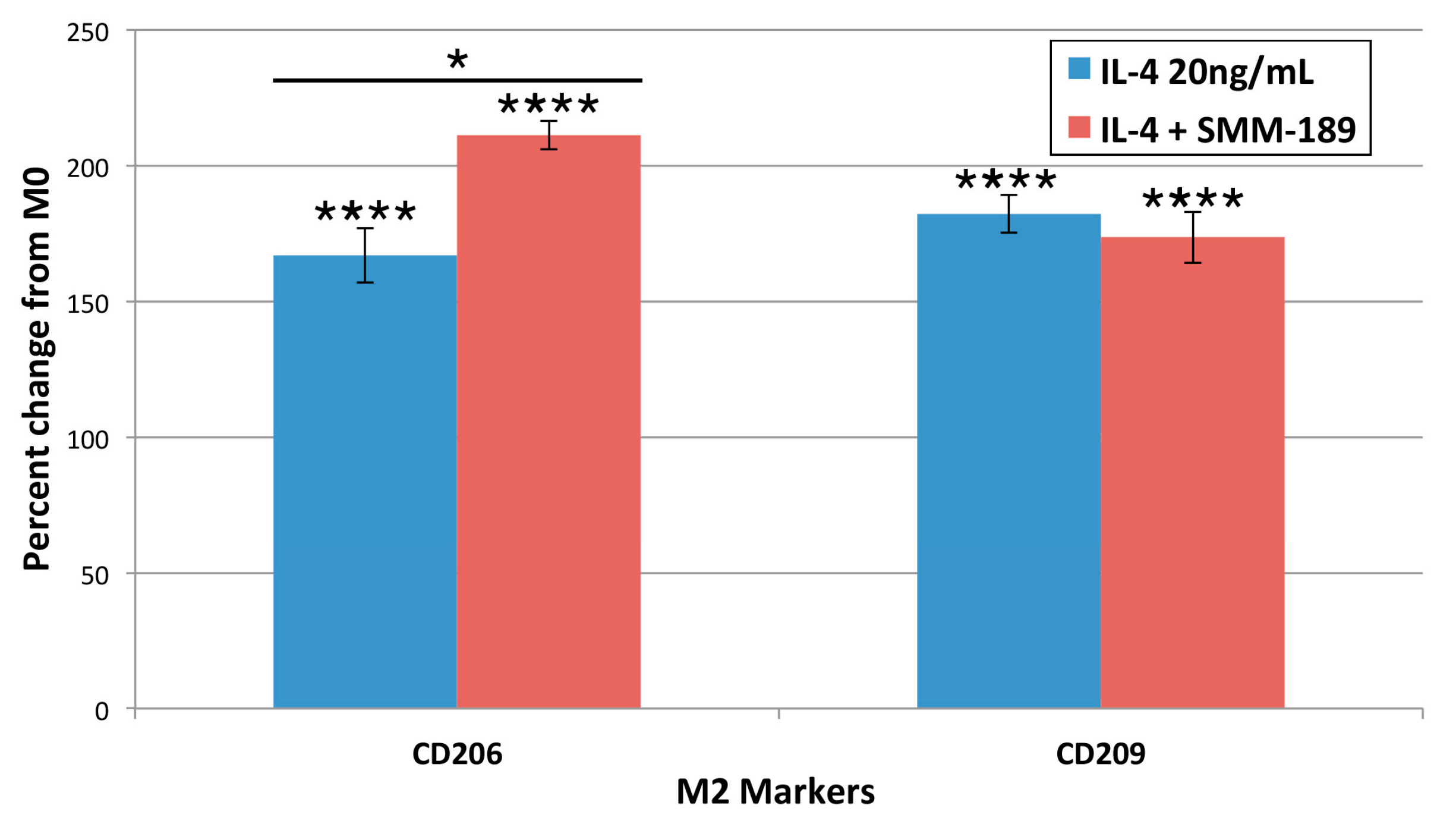
2.1.4. M2 Markers

2.1.5. Cannabinoid Type 2 Receptors (CB2)
2.2. Mice with Mild Traumatic Brain Injury (TBI)
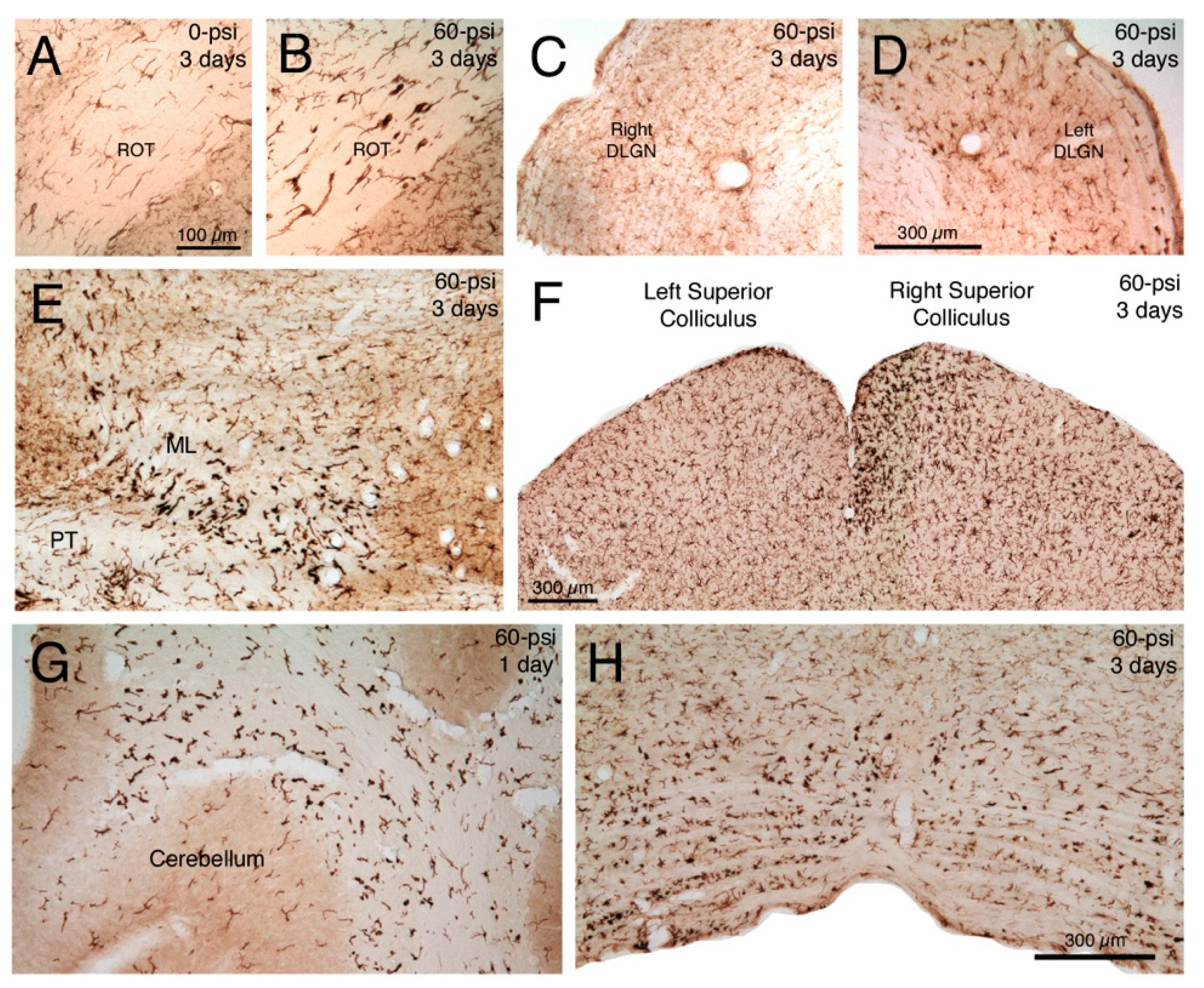
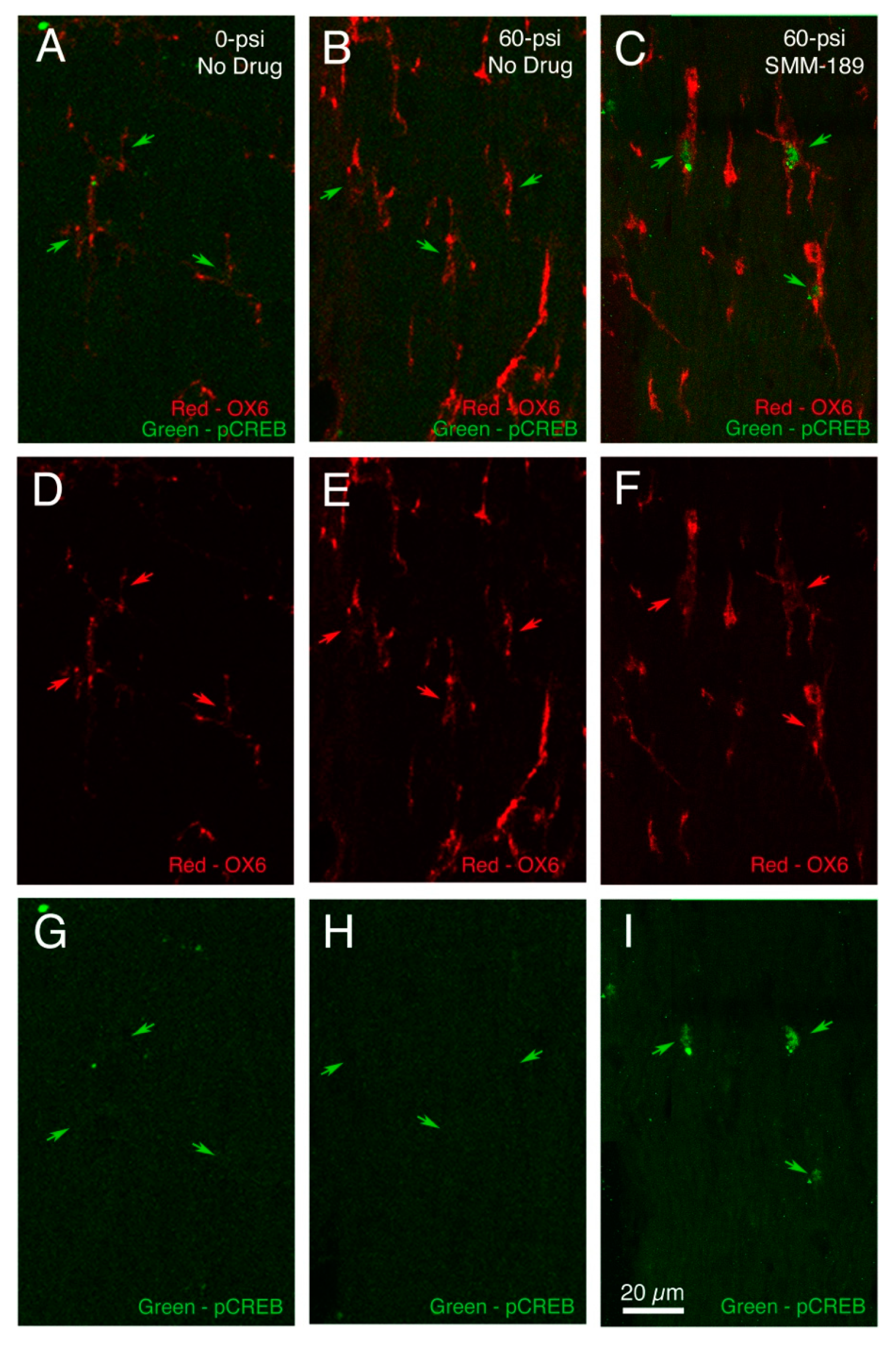
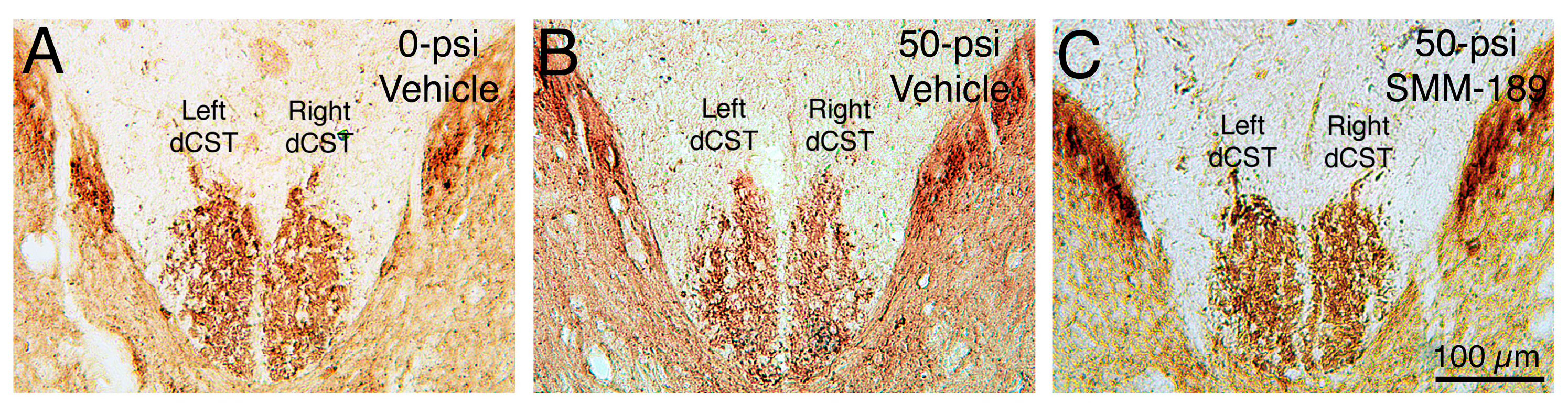
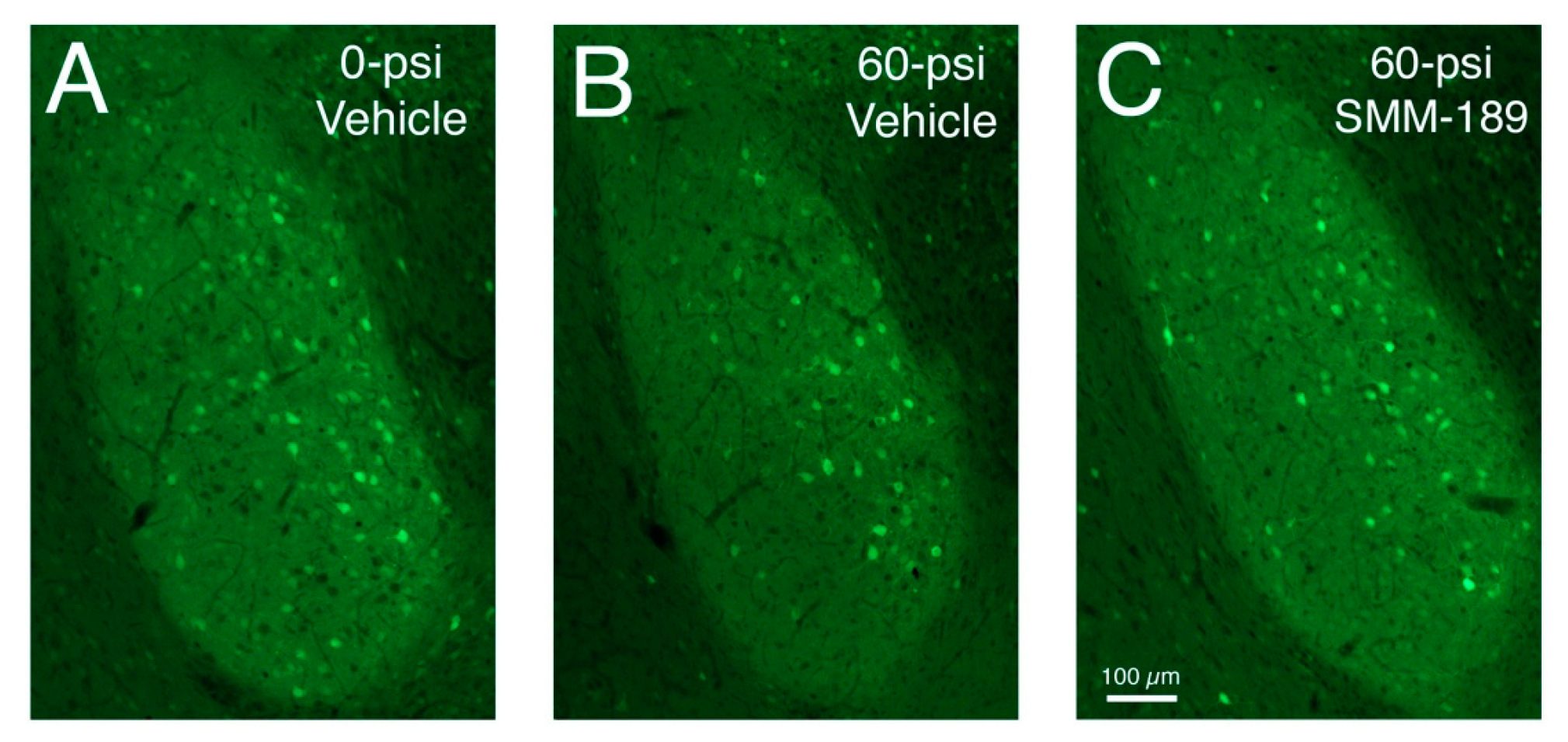
2.2.1. Microglial Responses to TBI and SMM-189


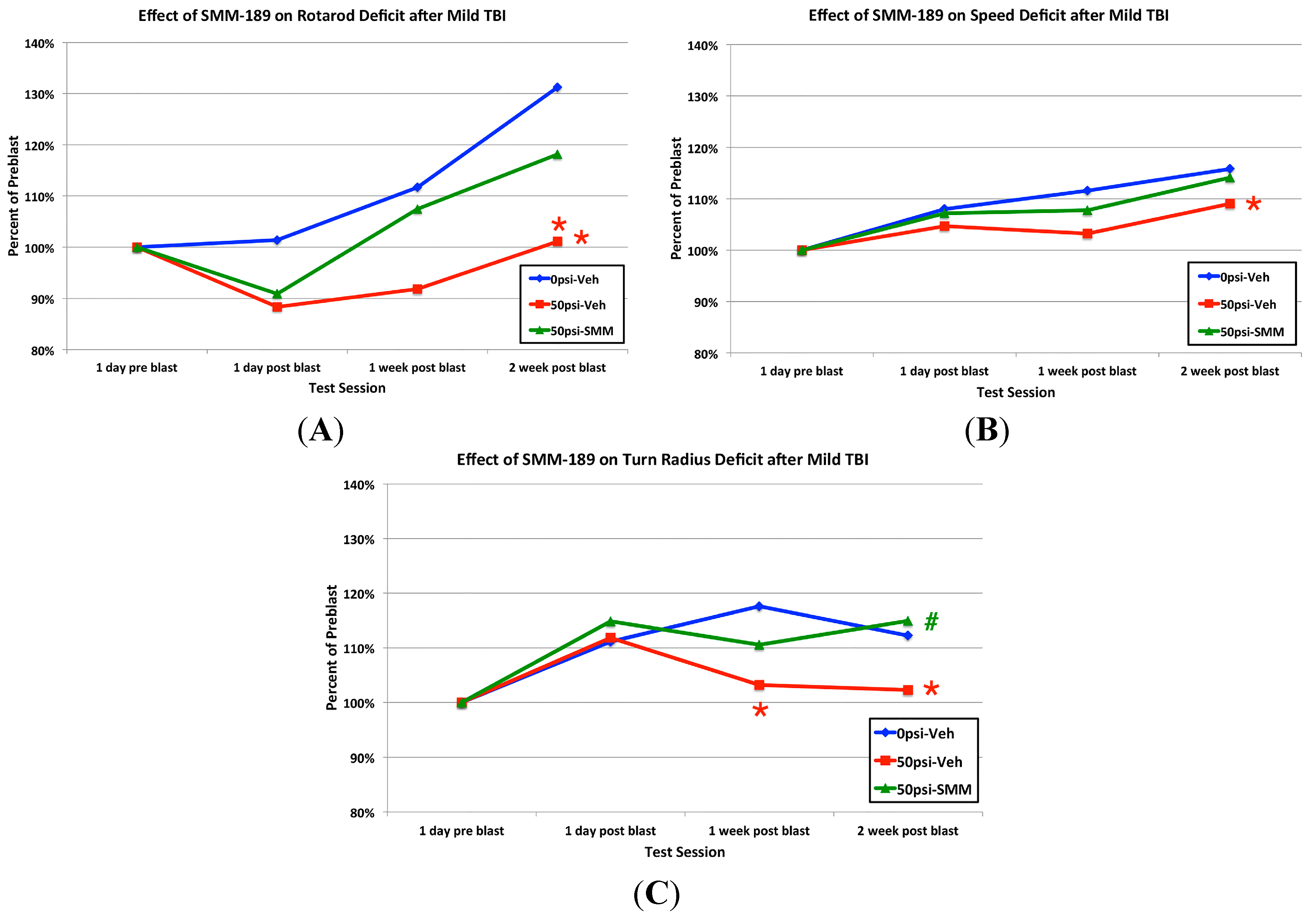
2.2.2. Motor Function


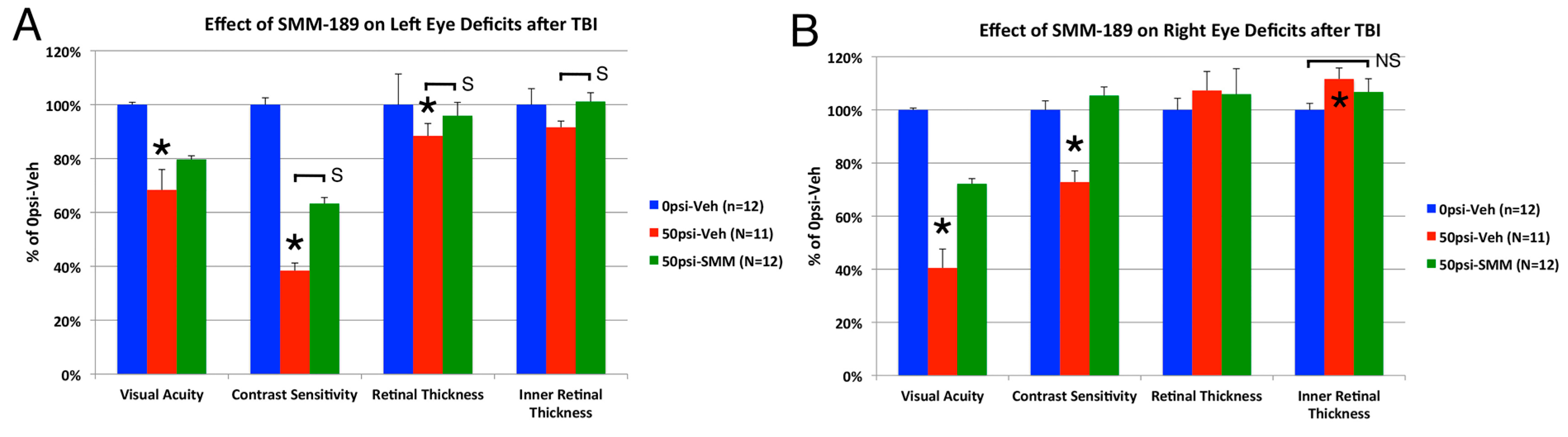
2.2.3. Visual Function

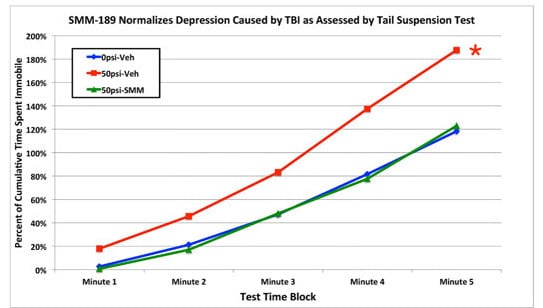
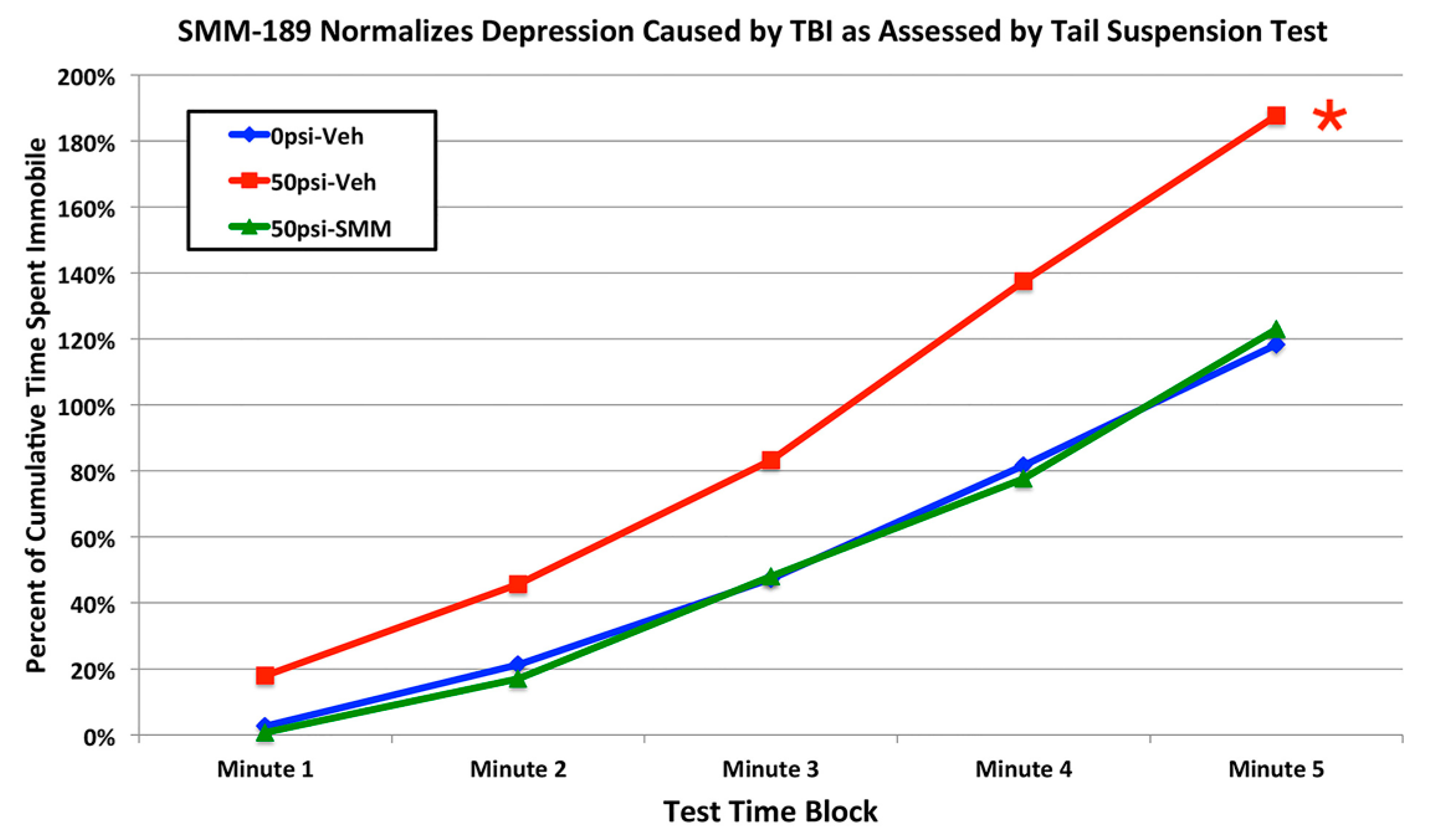
2.2.4. Depression and Fear



3. Discussion
3.1. Motor Deficits
3.2. Visual Deficits
3.3. Fear and Depression
3.4. Summary and Conclusions
4. Experimental Section
4.1. Studies of Human Microglia in Vitro
4.1.1. Cytokine and Chemokine Production by Primary Human Microglia
4.1.2. M1 versus M2 Marker Expression by Immortalized Human Microglia
4.2. Studies of Mild TBI in Mouse
4.2.1. Animals
4.2.2. TBI Methods
4.2.3. Behavioral Studies-Motor Assessment
4.2.4. Behavioral Studies-Visual Acuity and Contrast Sensitivity
4.2.5. Tail Suspension Depression Test
4.2.6. Fear Acquisition and Extinction Tests
4.3. Histological Studies
4.3.1. Tissue Fixation
4.3.2. Immunohistochemical Studies
4.3.3. Thy1-EYFP–Emx1-Cre Reporter Mice
4.3.4. Morphometry
4.4. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Risdall, J.E.; Menon, D.K. Traumatic brain injury. Phil. Trans. R. Soc. 2011, 366, 241–250. [Google Scholar] [CrossRef]
- Faul, M.; Xu, L.; Wald, M.M.; Coronado, V.G. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths, 2002–2006; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2010.
- Johnson, V.E.; Stewart, W.; Smith, D.H. Axonal pathology in traumatic brain injury. Exper. Neurol. 2013, 246, 35–43. [Google Scholar] [CrossRef]
- Bazarian, J.J.; Donnelly, K.; Peterson, D.R.; Warner, G.C.; Zhu, T.; Zhong, J. The relation between posttraumatic stress disorder and mild traumatic brain injury acquired during operations enduring freedom and Iraqi freedom. J. Head Trauma Rehabil. 2013, 28, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Namjoshi, D.R.; Good, C.; Cheng, W.H.; Panenka, W.; Richards, D.; Cripton, P.A.; Wellington, C.L. Towards clinical management of traumatic brain injury: A review of models and mechanisms from a biomechanical perspective. Dis. Models Mech. 2013, 6, 1325–1338. [Google Scholar] [CrossRef]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef]
- Kelley, B.J.; Lifshitz, J.; Povlishock, J.T. Neuroinflammatory responses after experimental diffuse traumatic brain injury. J. Neuropathol. Exp. Neurol. 2007, 66, 989–1001. [Google Scholar] [CrossRef]
- Redell, J.B.; Dash, P.K. Traumatic brain injury stimulates hippocampal catechol-O-methyl transferase expression in microglia. Neurosci. Lett. 2007, 413, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Thomas, T.C.; Ziebell, J.M.; Pauly, J.R.; Lifshitz, J. Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience 2012, 225, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Mohapatra, S.; Mohapatra, S.S. New perspectives on central and peripheral immune responses to acute traumatic brain injury. J. Neuroinflamm. 2012, 9, 236–247. [Google Scholar] [CrossRef]
- Kumar, A.; Loane, D.J. Neuroinflammation after traumatic brain injury: Opportunities for therapeutic intervention. Brain Behav. Immun. 2012, 26, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Patterson, Z.R.; Holahan, M.R. Understanding the neuroinflammatory response following concussion to develop treatment strategies. Front. Cell. Neurosci. 2012, 6, 58–90. [Google Scholar] [CrossRef] [PubMed]
- Perez-Polo, J.R.; Rea, H.C.; Johnson, K.M.; Parsley, M.A.; Unabia, G.C.; Xu, G.; Infante, S.K.; Dewitt, D.S.; Hulsebosch, C.E. Inflammatory consequences in a rodent model of mild traumatic brain injury. J. Neurotrauma 2013, 30, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Gentleman, S.M.; Leclercq, P.D.; Murray, L.S.; Griffin, W.S.; Graham, D.I.; Nicoll, J.A. The neuroinflammatory response in humans after traumatic brain injury. Neuropathol. Appl. Neurobiol. 2013, 39, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Heldt, S.A.; Elberger, A.J.; Deng, Y.; Guley, N.H.; del Mar, N.; Rogers, J.; Choi, G.W.; Ferrell, J.; Rex, T.S.; Honig, M.G.; et al. A novel closed-head model of mild traumatic brain injury caused by primary overpressure blast to the cranium produces sustained emotional deficits in mice. Front. Neurol. 2014, 5, 2–35. [Google Scholar] [CrossRef] [PubMed]
- Rex, T.; Elberger, A.J.; Deng, Y.; Guley, N.H.; D’Surney, L.; Hines-Beard, J.; del Mar, N.; Honig, M.G.; Reiner, A. Visual deficits in mice after mild traumatic brain injury produced using a novel closed-head model of primary overpressure blast. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5095–B0053. [Google Scholar]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef]
- Schomberg, D.; Olson, J. Immune responses of microglia in the spinal cord: Contribution to pain states. Exp. Neurol. 2012, 234, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Benito, C.; Núñez, E.; Tolón, RM.; Carrier, EJ.; Rábano, A.; Hillard, CJ.; Romero, J. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J. Neurosci. 2003, 23, 11136–11141. [Google Scholar]
- Stella, N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes and astrocytomas. Glia 2010, 58, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Darlington, C.L.; Smith, P.F.; Ashton, J.C. Antibody testing for brain immunohistochemistry: Brain immunolabeling for the cannabinoid CB2 receptor. J. Neurosci. Methods 2013, 216, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Ashton, J.C.; Glass, M. The cannabinoid CB2 receptor as a target for inflammation-dependent neurodegeneration. Curr. Neuropharmacol. 2007, 5, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Romero-Sandoval, E.A.; Horvath, R.; Landry, R.P.; DeLeo, J.A. Cannabinoid receptor type 2 activation induces a microglial anti-inflammatory phenotype and reduces migration via MKP induction and ERK dephosphorylation. Mol. Pain 2009, 5, 25. [Google Scholar] [CrossRef]
- Arévalo-Martín, A.; Vela, J.M.; Molina-Holgado, E.; Borrell, J.; Guaza, C. Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J. Neurosci. 2003, 23, 2511–2516. [Google Scholar] [PubMed]
- Palazuelos, J.; Aguado, T.; Pazos, M.R.; Julien, B.; Carrasco, C.; Resel, E.; Sagredo, O.; Benito, C.; Romero, J.; Azcoitia, I.; et al. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington’s disease excitotoxicity. Brain 2009, 132, 3152–3164. [Google Scholar] [CrossRef] [PubMed]
- Rivers, J.R.J.; Ashton, J.C. The development of cannabinoid CBII receptor agonists for the treatment of central neuropathies. Cent. Nerv. Syst. Agents Med. Chem. 2010, 10, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, W.; Schäfer, F.; Striggow, V.; Fröhlich, K.; Striggow, F. Cannabinoid receptor subtypes 1 and 2 mediate long-lasting neuroprotection and improve motor behavior deficits after transient focal cerebral ischemia. Neuroscience 2012, 227, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Spatz, M.; Shohami, E. Endocannabinoids and neuroprotection. Science 2002, 2002, re5. [Google Scholar] [CrossRef]
- Meyer, M.J.; Megyesi, J.; Meythaler, J.; Murie-Fernandez, M.; Aubut, J.A.; Foley, N.; Salter, K.; Bayley, M.; Marshall, S.; Teasell, R. Acute management of acquired brain injury part II: An evidence-based review of pharmacological interventions. Brain Inj. 2010, 24, 706–721. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.B.; Tuma, R.F.; Amenta, P.S.; Barbe, M.F.; Jallo, J.I. Acute effects of a selective cannabinoid-2 receptor agonist on neuroinflammation in a model of traumatic brain injury. J. Neurotrauma 2011, 28, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Amenta, P.S.; Jallo, J.I.; Tuma, R.F.; Elliott, M.B. A cannabinoid type-2 receptor agonist attenuates blood-brain barrier damage and neurodegeneration in a murine model of traumatic brain injury. J. Neurosci. Res. 2012, 90, 2293–2305. [Google Scholar] [CrossRef] [PubMed]
- Firsching, R.; Piek, J.; Skalej, M.; Rohde, V.; Schmidt, U.; Striggow, F.; KN38–7271 Study Group. Early survival of comatose patients after severe traumatic brain injury with the dual cannabinoid CB1/CB2 receptor agonist KN38–7271: A randomized, double-blind, placebo-controlled phase II trial. J. Neurol. Surg. 2012, 73, 204–216. [Google Scholar] [CrossRef]
- Koppel, J.; Vingtdeux, V.; Marambaud, P.; D’Abramo, C.; Jimenez, H.; Stauber, M.; Friedman, R.; Davies, P. CB2 receptor deficiency increases amyloid pathology and alters tau processing in a transgenic mouse model of Alzheimer’s disease. Mol. Med. 2013, 20, 29–36. [Google Scholar]
- Lopez-Rodriguez, A.B.; Siopi, E.; Finn, D.P.; Marchand-Leroux, C.; Garcia-Segura, L.M.; Jafarian-Tehrani, M.; Viveros, M.P. CB1 and CB2 cannabinoid receptor antagonists prevent minocycline-induced neuroprotection following traumatic brain injury in mice. Cereb. Cortex 2013. [Google Scholar] [CrossRef]
- Lunn, C.; Fine, J.; Rojas-Triana, A.; Jackson, J.; Fan, X.D.; Kung, T.; Gonsiorek, W.; Schwarz, M.; Lavey, B.; Kozlowski, J.; et al. A novel cannabinoid peripheral cannabinoid receptor-selective inverse agonist blocks leukocyte recruitment in vivo. J. Pharmacol. Exp. Ther. 2006, 316, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Lunn, C.; Reich, E.P.; Fine, J.; Lavey, B.; Kozlowski, J.; Hipkin, R.; Lundell, D.; Bober, L. Biology and therapeutic potential of cannabinoid CB2 receptor inverse agonists. Br. J. Pharmacol. 2008, 153, 226–239. [Google Scholar] [CrossRef]
- Atwood, B.K.; Straiker, A.; Mackie, K. CB2: Therapeutic target-in-waiting. Prog. Neuro-Psychopharmacol. Biol. Psychiat. 2012, 38, 16–20. [Google Scholar] [CrossRef]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, H.; Gurley, S.N.; Moore, B.M. Design and synthesis of novel tri-aryl CB2 selective cannabinoid ligands. Bioorg. Med. Chem. Lett. 2009, 19, 1691–1693. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, M.; Kumata, K.; Yanamoto, K.; Kawamura, K.; Yamasaki, T.; Yui, J.; Hatori, A.; Ogawa, M.; Yoshida, Y.; Nengaki, N.; et al. Radiosynthesis of novel carbon-11-labeled triaryl ligands for cannabinoid-type 2 receptor. Bioorg. Med. Chem. Lett. 2010, 20, 1565–1568. [Google Scholar] [CrossRef] [PubMed]
- Tang-Schomer, M.D.; Patel, A.R.; Baas, P.W.; Smith, D.H. Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEB J. 2010, 24, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Tang-Schomer, M.D.; Johnson, V.E.; Baas, P.W.; Stewart, W.; Smith, D.H. Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Exp. Neurol. 2012, 233, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Love, S.; Louis, D.N.; Ellison, D.W. Trauma. In Greenfield’s Neuropathology, 8th ed.; Edward Arnold Publishers Ltd.: London, UK, 2008; pp. 733–812. [Google Scholar]
- Smith, J.; Das, A.; Ray, S.; Banik, N. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Petras, J.; Bauman, R.; Elsayed, N. Visual system degeneration induced by blast overpressure. Toxicology 1997, 121, 41–49. [Google Scholar] [CrossRef]
- Koliatsos, V.E.; Cernak, I.; Xu, L.; Song, Y.; Savonenko, A.; Crain, B.J.; Eberhart, C.G.; Frangakis, C.E.; Melnikova, T.; Kim, H.; et al. A mouse model of blast injury to brain: Initial pathological, neuropathological, and behavioral characterization. J. Neuropathol. Exp. Neurol. 2011, 70, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hamm, R.J.; Povlishock, J.T. Traumatic axonal injury in the optic nerve: Evidence for axonal swelling, disconnection, dieback, and reorganization. J. Neurotrauma 2011, 28, 1185–1198. [Google Scholar] [CrossRef]
- Mohan, K.; Kecova, H.; Hernandez-Merino, E.; Kardon, R.H.; Harper, M.M. Retinal ganglion cell damage in an experimental rodent model of blast-mediated traumatic brain injury. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3440–3450. [Google Scholar] [CrossRef]
- Tzekov, R.; Quezada, A.; Gautier, M.; Biggins, D.; Frances, C.; Mouzon, B.; Jamison, J.; Mullan, M.; Crawford, F. Repetitive mild traumatic brain injury causes optic nerve and retinal damage in a mouse model. J. Neuropathol. Exp. Neurol. 2014, 73, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Cockerham, G.C.; Goodrich, G.L.; Weichel, E.D.; Orcutt, J.C.; Rizzo, J.F.; Bower, K.S. Schuchard, R.A. Eye and visual function in traumatic brain injury. J. Rehabil. Res. Dev. 2009, 46, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Warner, N.; Eggenberger, E. Traumatic optic neuropathy: A review of the current literature. Curr. Opin. Ophthalmol. 2010, 21, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Peskind, P.R.; Brody, D.; Cernak, I.; McKee, A.; Ruff, R.L. Military- and sports-related mild traumatic brain injury: Clinical presentation, management, and long-term consequences. J. Clin. Psychiatry 2013, 74, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Di Leo, M.A.; Caputo, S.; Falsini, B.; Porciatti, V.; Minnella, A.; Greco, A.V.; Ghirlanda, G. Nonselective loss of contrast sensitivity in visual system testing in early type I diabetes. Diabetes Care 1992, 15, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Hasegawa, S.; Takagi, M.; Yoshizawa, T.; Usui, T. Contrast sensitivity for the stationary and drifting vertical stripe patterns in patients with optic nerve disorders. Ophthalmalogica 1993, 207, 100–105. [Google Scholar] [CrossRef]
- Hashemi, H.; Khabazkhoob, M.; Jafarzadehpur, E.; Emamian, M.H.; Shariati, M.; Fotouhi, A. Contrast sensitivity evaluation in a population-based study in Shahroud, Iran. Ophthalmology 2012, 119, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.F. Contrast sensitivity versus visual acuity in retinal disease. Br. J. Ophthalmol. 1986, 70, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Al-Hashmi, A.M.; Kramer, D.J.; Mullen, K.T. Human vision with a lesion of the parvocellular pathway: An optic neuritis model for selective contrast sensitivity deficits with severe loss of midget ganglion cell function. Exp. Brain Res. 2011, 215, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Akimov, N.P.; Renteria, R.C. Spatial frequency threshold and contrast sensitivity of an optomotor behavior are impaired in the Ins2Akita mouse model of diabetes. Behav. Brain Res. 2012, 226, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Dräger, U.C.; Olsen, J.F. Origins of crossed and uncrossed retinal projections in pigmented and albino mice. J. Comp. Neurol. 1980, 191, 383–412. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, M.; Bobić-Radovanović, A.; Vuković, D.; Knezević, M.; Risović, D. Ocular injuries caused by airsoft guns: Ten-year experience. Acta Chir Iugosl. 2012, 59, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Hines-Beard, J.; Marchetta, J.; Gordon, S.; Chaum, E.; Geisert, E.E.; Rex, T.S. A novel mouse model of ocular blast injury. Exp. Eye Res. 2012, 99, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Jasnow, A.M.; Ehrlich, D.E.; Choi, D.C.; Dabrowska, J.; Bowers, M.E.; McCullough, K.M.; Rainnie, D.G.; Ressler, K.J. Thy1-expressing neurons in the basolateral amygdala may mediate fear inhibition. J. Neurosci. 2013, 33, 10396–10404. [Google Scholar] [CrossRef] [PubMed]
- Herry, C.; Ciocchi, S.; Senn, V.; Demmou, L.; Müller, C.; Lüthi, A. Switching on and off fear by distinct neuronal circuits. Nature 2008, 454, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Gorski, J.A.; Talley, T.; Qiu, M.; Puelles, L.; Rubenstein, J.L.R.; Jones, K.R. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the emx1-expressing lineage. J. Neurosci. 2002, 22, 6309–6314. [Google Scholar] [PubMed]
- Bareyre, F.M.; Kerschensteiner, M.; Misgeld, T.; Sanes, J.R. Transgenic labeling of the corticospinal tract for monitoring axonal responses to spinal cord injury. Nat. Med. 2005, 11, 1355–1360. [Google Scholar] [CrossRef] [PubMed]
- Drai, D.; Golani, I. SEE: A tool for the visualization and analysis of rodent exploratory behavior. Neurosci. Biobehav. Rev. 2001, 25, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Drai, D.; Benjamini, Y.; Golani, I. Statistical discrimination of natural modes of motion in rat exploratory behavior. J. Neurosci. Methods 2000, 96, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Kafkafi, N.; Lipkind, D.; Benjamini, Y.; Mayo, C.L.; Elmer, G.I.; Golani, I. See locomotor behavior test discriminates C57BL/6J and DBA/2J mouse inbred strains across laboratories and protocol conditions. Behav. Neurosci. 2003, 117, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Kafkafi, N.; Mayo, C.; Drai, D.; Golani, I.; Elmer, G. Natural segmentation of the locomotor behavior of drug-induced rats in a photobeam cage. J. Neurosci. Methods 2001, 109, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Lipkind, D.; Sakov, A.; Kafkafi, N.; Elmer, G.I.; Benjamini, Y.; Golani, I. New replicable anxiety-related measures of wall versus center behavior of mice in the open field. J. Appl. Physiol. 2004, 97, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Lafferty, D.C.; Wang, H.B.; del Mar, N.; Deng, Y.P. The group 2 metabotropic glutamate receptor agonist LY379268 rescues neuronal, neurochemical and motor abnormalities in R6/2 Huntington’s disease mice. Neurobiol. Dis. 2012, 47, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Prusky, G.T.; Alam, N.M.; Beekman, S.; Douglas, R.M. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4611–4616. [Google Scholar] [CrossRef]
- Rao, V.; Bertrand, M.; Rosenberg, P.; Makley, M.; Schretlen, D.J.; Brandt, J.; Mielke, M.M. Predictors of new-onset depression after mild traumatic brain injury. J. Neuropsychiatry Clin. Neurosci. 2010, 22, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; Mombereaqu, C.; Vassout, A. The tail suspension test as a model for assessing antidepressant activity: Review of pharmacological and genetic studies in mice. Neurosci. Biobehav. Res. 2005, 29, 571–625. [Google Scholar] [CrossRef]
- Mahan, A.L.; Ressler, K.J. Fear conditioning, synaptic plasticity and the amygdala: Implications for posttraumatic stress disorder. Trends Neurosci. 2012, 35, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, S.; LeDoux, M.S.; Jones, S.; Jones, J.; Zhou, Q.; Gong, S.; Ryan, P.; Reiner, A. Localization of preganglionic neurons that innervate choroidal neurons of pterygopalatine ganglion. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3713–3724. [Google Scholar] [CrossRef]
- Meade, C.A.; Figueredo-Cardenas, G.; Fusco, F.R.; Nowak, T.S.; Pulsinelli, W.; Reiner, A. Transient global ischemia in rats yields striatal projection neuron and interneuron loss resembling that in Huntington’s disease. Exper. Neurol. 2000, 166, 307–323. [Google Scholar] [CrossRef]
- Shitaka, Y.; Tran, H.T.; Bennett, R.E.; Sanchez, L.; Levy, M.A.; Dikranian, K.; Brody, D.L. Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J. Neuropathol. Exp. Neurol. 2011, 70, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Ebneter, A.; Casson, R.J.; Wood, J.P.; Chidlow, G. Microglial activation in the visual pathway in experimental glaucoma: Spatiotemporal characterization and correlation with axonal injury. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6448–6460. [Google Scholar] [CrossRef]
- Acosta, S.A.; Tajiri, N.; Shinozuka, K.; Ishikawa, H.; Grimmig, B.; Diamond, D.M.; Sanberg, P.R.; Bickford, P.C.; Kaneko, Y.; Borlongan, C.V. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PLoS One 2013, 8, e53376. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, E.J.; Moon, L.D.; Popat, R.J.; King, V.R.; Bennett, G.S.; Patel, P.N.; Fawcett, J.W.; McMahon, S.B. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 2002, 416, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Shirai, Y. Protein kinase Cγ (PKCγ): Function of neuron specific isotype. J. Biochem. 2002, 132, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Lieu, A.; Tenorio, G.; Kerr, B.J. Protein kinase C gamma (PKCγ) as a novel marker to assess the functional status of the corticospinal tract in experimental autoimmune encephalomyelitis (EAE). J. Neuroimmunol. 2013, 256, 43–48. [Google Scholar] [CrossRef]
- Deng, Y.P.; Wong, T.; Bricker-Anthony, C.; Deng, B.; Reiner, A. Loss of corticostriatal and thalamostriatal synaptic terminals in heterozygous Q140 Huntington’s disease mice. Neurobiol. Dis. 2013, 60, 89–107. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; del Mar, N.; Deng, Y.P.; Meade, C.A.; Sun, Z.; Goldowitz, D. R6/2 Neurons with intranuclear inclusions survive for prolonged periods in the brains of chimeric mice. J. Comp. Neurol. 2007, 505, 603–629. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reiner, A.; Heldt, S.A.; Presley, C.S.; Guley, N.H.; Elberger, A.J.; Deng, Y.; D'Surney, L.; Rogers, J.T.; Ferrell, J.; Bu, W.; et al. Motor, Visual and Emotional Deficits in Mice after Closed-Head Mild Traumatic Brain Injury Are Alleviated by the Novel CB2 Inverse Agonist SMM-189. Int. J. Mol. Sci. 2015, 16, 758-787. https://doi.org/10.3390/ijms16010758
Reiner A, Heldt SA, Presley CS, Guley NH, Elberger AJ, Deng Y, D'Surney L, Rogers JT, Ferrell J, Bu W, et al. Motor, Visual and Emotional Deficits in Mice after Closed-Head Mild Traumatic Brain Injury Are Alleviated by the Novel CB2 Inverse Agonist SMM-189. International Journal of Molecular Sciences. 2015; 16(1):758-787. https://doi.org/10.3390/ijms16010758
Chicago/Turabian StyleReiner, Anton, Scott A. Heldt, Chaela S. Presley, Natalie H. Guley, Andrea J. Elberger, Yunping Deng, Lauren D'Surney, Joshua T. Rogers, Jessica Ferrell, Wei Bu, and et al. 2015. "Motor, Visual and Emotional Deficits in Mice after Closed-Head Mild Traumatic Brain Injury Are Alleviated by the Novel CB2 Inverse Agonist SMM-189" International Journal of Molecular Sciences 16, no. 1: 758-787. https://doi.org/10.3390/ijms16010758
APA StyleReiner, A., Heldt, S. A., Presley, C. S., Guley, N. H., Elberger, A. J., Deng, Y., D'Surney, L., Rogers, J. T., Ferrell, J., Bu, W., Del Mar, N., Honig, M. G., Gurley, S. N., & II, B. M. M. (2015). Motor, Visual and Emotional Deficits in Mice after Closed-Head Mild Traumatic Brain Injury Are Alleviated by the Novel CB2 Inverse Agonist SMM-189. International Journal of Molecular Sciences, 16(1), 758-787. https://doi.org/10.3390/ijms16010758