
Regulation of Human Adenovirus Alternative RNA Splicing by the Adenoviral L4-33K and L4-22K Proteins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Adenovirus Alternative RNA Splicing

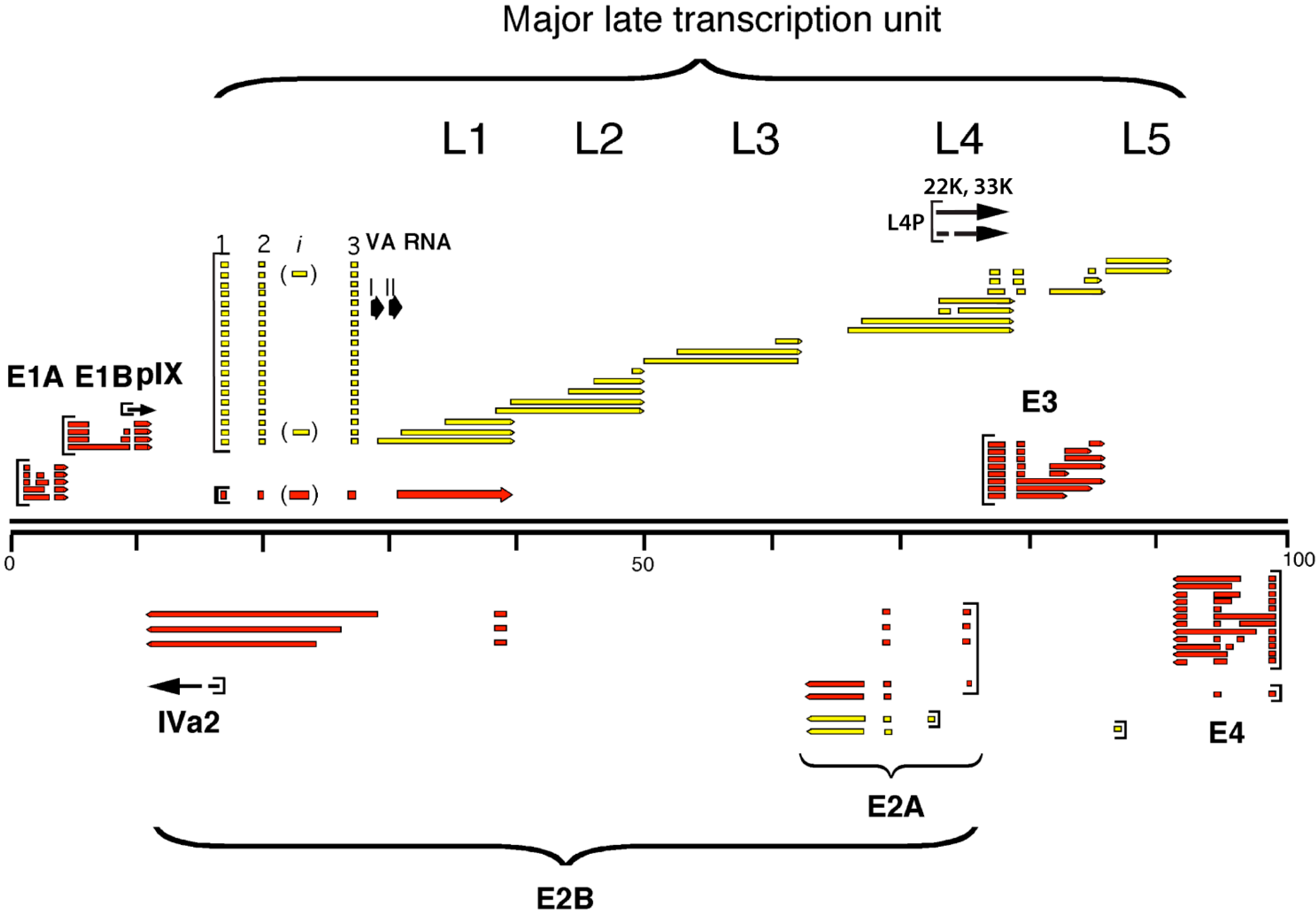
2.1. The Major Late Transcription Unit
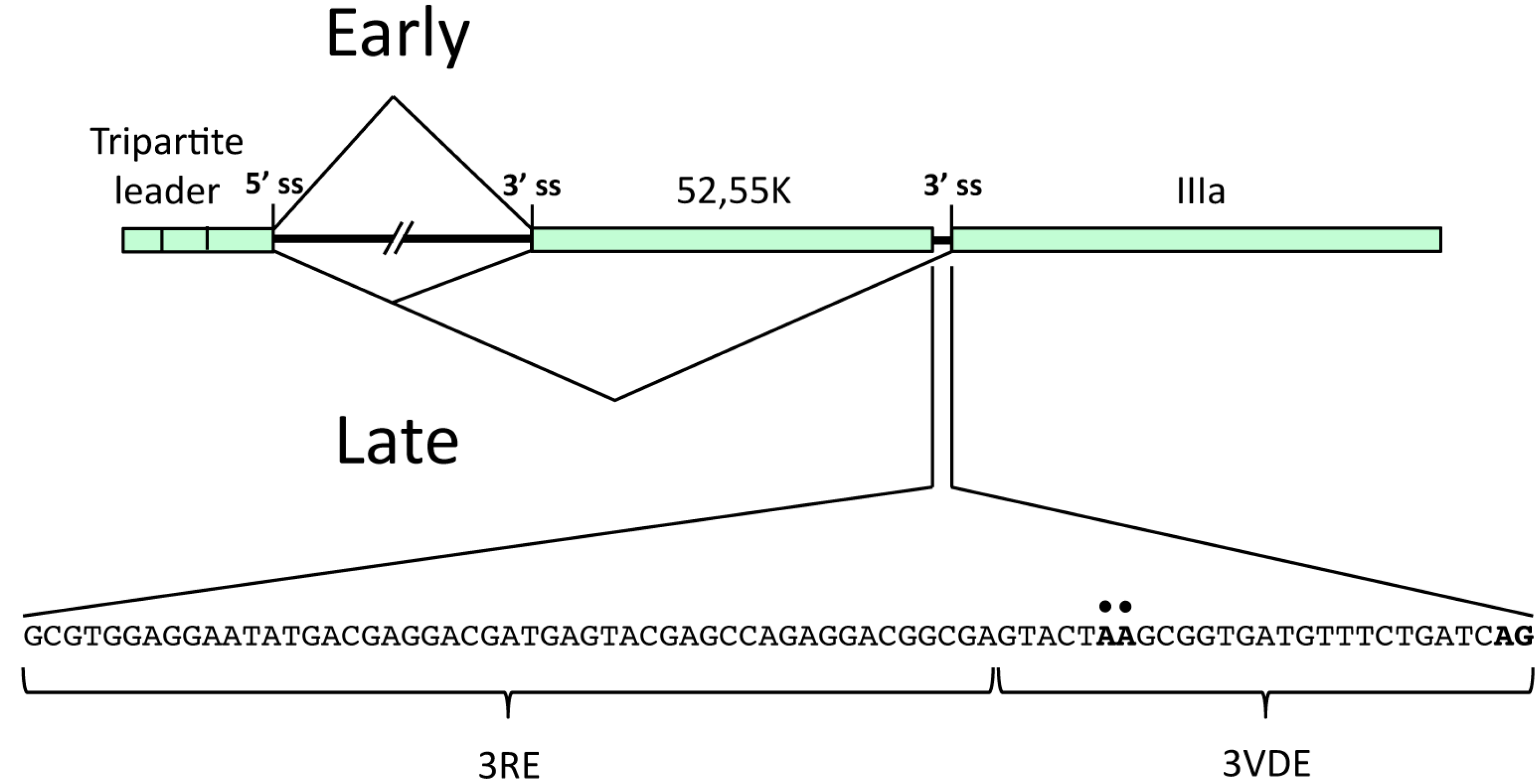
2.1.1. The L1 Model System

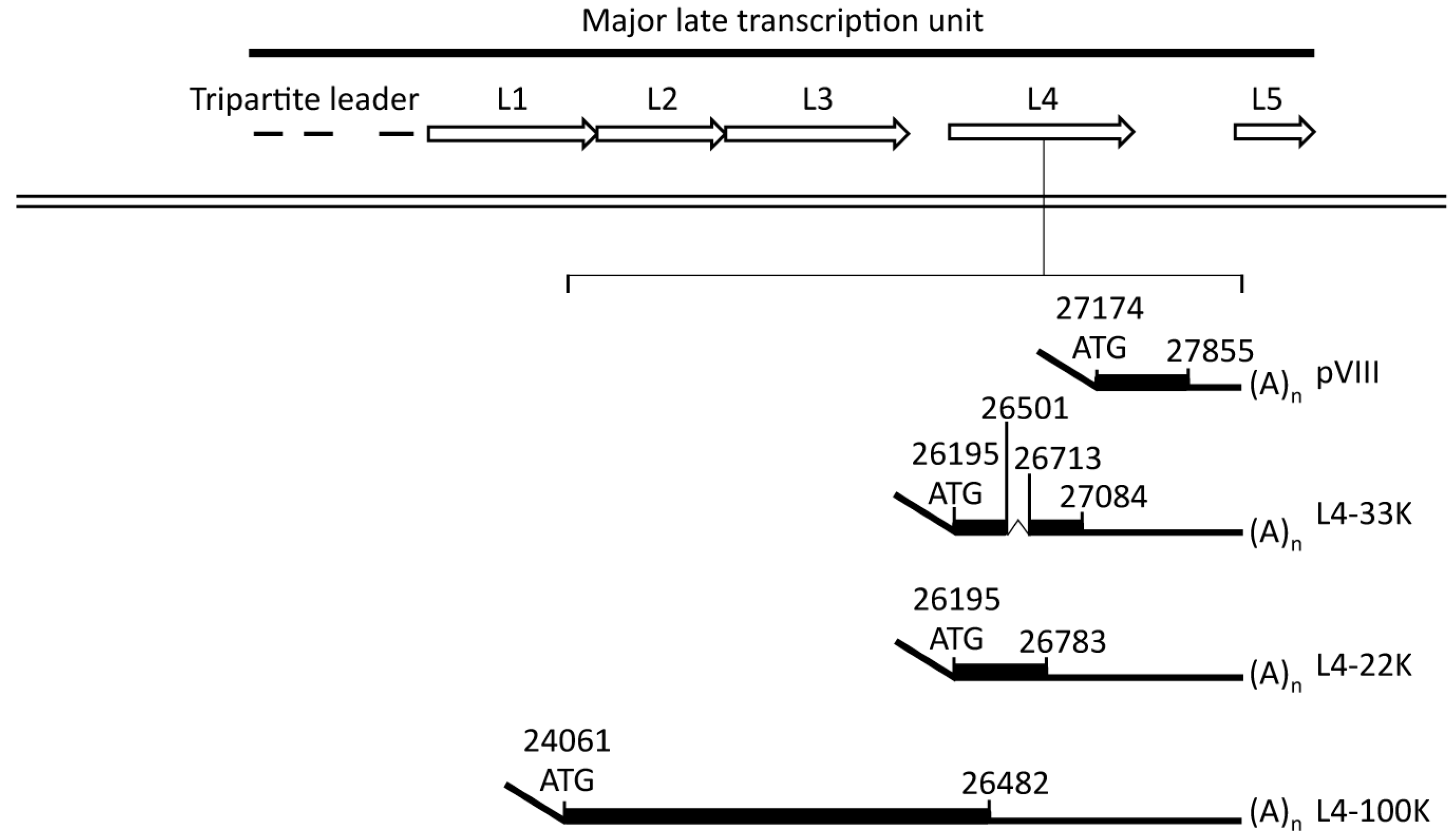
2.1.2. The L4 Transcript Proteins

2.2. L4-22K

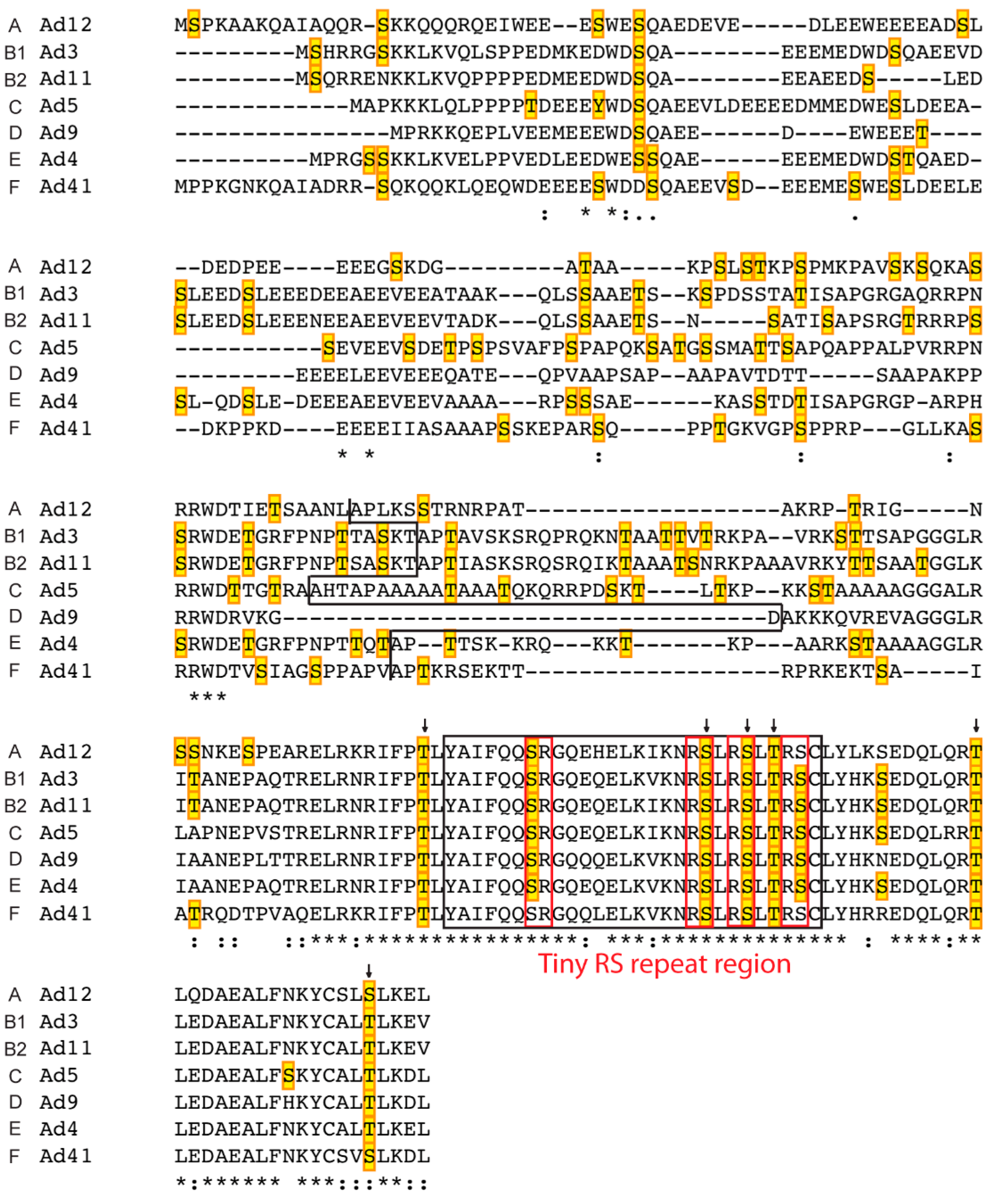
2.3. L4-33K

3. Post-Translational Modification of Adenoviral Proteins
3.1. A Janus Effect of Two Cellular Protein Kinases on the Activity of L4-33K as an Alternative Splicing Enhancer Protein
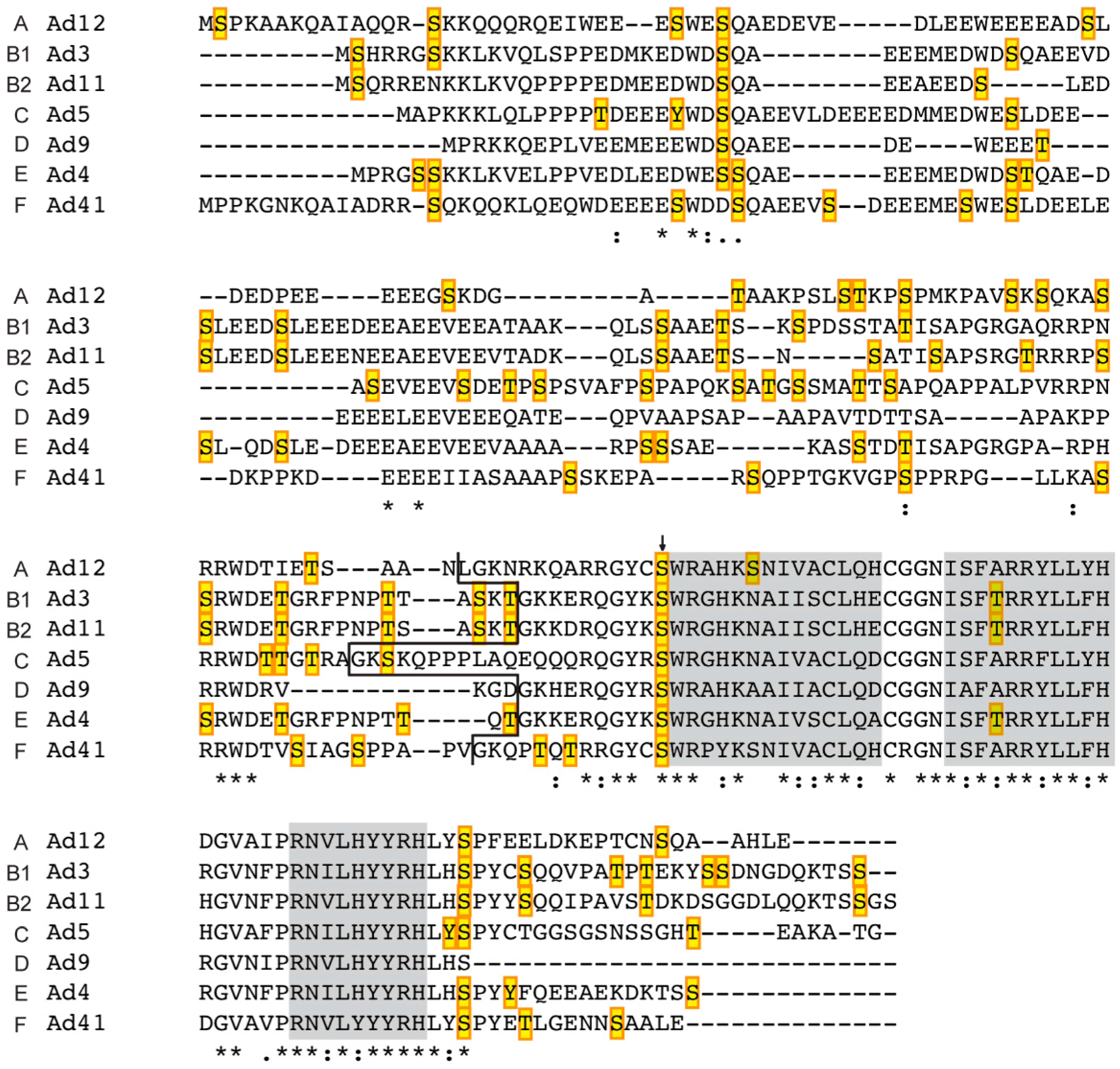
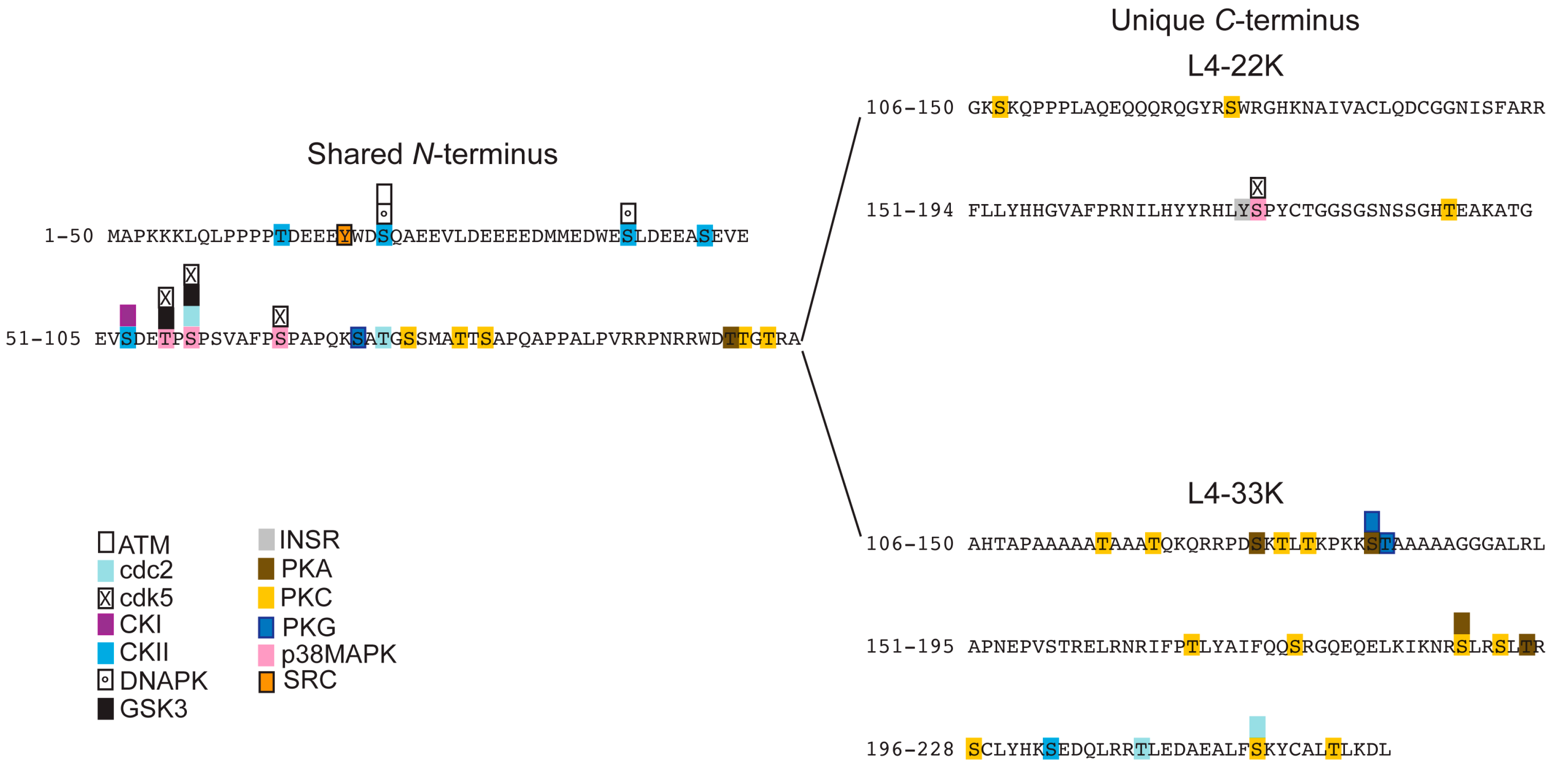
3.2. Prediction of Phosphorylation Sites in L4-33K and L4-22K

Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Berget, S.M.; Moore, C.; Sharp, P.A. Spliced segments at the 5' terminus of adenovirus 2 late mrna. Proc. Natl. Acad. Sci. USA 1977, 74, 3171–3175. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.T.; Gelinas, R.E.; Broker, T.R.; Roberts, R.J. An amazing sequence arrangement at the 5' ends of adenovirus 2 messenger RNA. Cell 1977, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Akusjarvi, G.; Stevenin, J. Remodelling of the host cell rna splicing machinery during an adenovirus infection. Curr. Top. Microbiol. Immunol. 2003, 272, 253–286. [Google Scholar] [PubMed]
- Wold, W.S.; Tollefson, A.E.; Hermiston, T.W. E3 transcription unit of adenovirus. Curr. Top. Microbiol. Immunol. 1995, 199, 237–274. [Google Scholar] [PubMed]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Lion, T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin. Microbiol. Rev. 2014, 27, 441–462. [Google Scholar] [CrossRef] [PubMed]
- Akusjarvi, G. Temporal regulation of adenovirus major late alternative RNA splicing. Front. Biosci. 2008, 13, 5006–5015. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Benko, M.; Harrach, B. Genetic content and evolution of adenoviruses. J. Gen. Virol. 2003, 84, 2895–2908. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chen, M.; Pettersson, U. A new look at adenovirus splicing. Virology 2014, 456–457, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Kvissel, A.-K.; Persson, H.T.; Aksaas, A-K.; Akusjärvi, G.; Skålhegg, B.S. Phosphorylation-dependent regulation of adenovirus alternative RNA splicing by PKA, DNA-PK, PP2A and SR proteins. In Virology II Advanced issues; iConcept Press: Hong Kong, China, 2014; pp. 257–287. [Google Scholar]
- Young, C.S. The structure and function of the adenovirus major late promoter. Curr. Top. Microbiol. Immunol. 2003, 272, 213–249. [Google Scholar] [PubMed]
- Shaw, A.R.; Ziff, E.B. Transcripts from the adenovirus-2 major late promoter yield a single early family of 3' coterminal mrnas and five late families. Cell 1980, 22, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, S.; Eggerding, F.; Falck-Pederson, E.; Darnell, J.E., Jr. Transcription unit mapping in adenovirus: Regions of termination. J. Virol. 1986, 59, 112–119. [Google Scholar] [PubMed]
- Chow, L.T.; Broker, T.R.; Lewis, J.B. Complex splicing patterns of rnas from the early regions of adenovirus-2. J. Mol. Biol. 1979, 134, 265–303. [Google Scholar] [CrossRef] [PubMed]
- Akusjarvi, G.; Persson, H. Controls of rna splicing and termination in the major late adenovirus transcription unit. Nature 1981, 292, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Nevins, J.R.; Wilson, M.C. Regulation of adenovirus-2 gene expression at the level of transcriptional termination and rna processing. Nature 1981, 290, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.; Svensson, C.; Akusjarvi, G. Control of adenovirus major late gene expression at multiple levels. J. Mol. Biol. 1992, 225, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.J.; Leppard, K.N. Adenovirus serotype 5 l4–22k and l4–33k proteins have distinct functions in regulating late gene expression. J. Virol. 2009, 83, 3049–3058. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.J.; Scott, G.E.; Leppard, K.N. Adenovirus late-phase infection is controlled by a novel l4 promoter. J. Virol. 2010, 84, 7096–7104. [Google Scholar] [CrossRef] [PubMed]
- Hasson, T.B.; Soloway, P.D.; Ornelles, D.A.; Doerfler, W.; Shenk, T. Adenovirus l1 52- and 55-kilodalton proteins are required for assembly of virions. J. Virol. 1989, 63, 3612–3621. [Google Scholar] [PubMed]
- Gustin, K.E.; Imperiale, M.J. Encapsidation of viral DNA requires the adenovirus L1 52/55-kilodalton protein. J. Virol. 1998, 72, 7860–7870. [Google Scholar] [PubMed]
- San Martin, C. Latest insights on adenovirus structure and assembly. Viruses 2012, 4, 847–877. [Google Scholar]
- Kanopka, A.; Muhlemann, O.; Akusjarvi, G. Inhibition by SR proteins of splicing of a regulated adenovirus pre-mRNA. Nature 1996, 381, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Dauksaite, V.; Akusjarvi, G. Human splicing factor ASF/SF2 encodes for a repressor domain required for its inhibitory activity on pre-mRNA splicing. J. Biol. Chem. 2002, 277, 12579–12586. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.S.; Nilsson, C.E.; Punga, T.; Akusjarvi, G. Functional inactivation of the SR family of splicing factors during a vaccinia virus infection. EMBO Rep. 2002, 3, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Kanopka, A.; Muhlemann, O.; Petersen-Mahrt, S.; Estmer, C.; Ohrmalm, C.; Akusjarvi, G. Regulation of adenovirus alternative RNA splicing by dephosphorylation of SR proteins. Nature 1998, 393, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Estmer Nilsson, C.; Petersen-Mahrt, S.; Durot, C.; Shtrichman, R.; Krainer, A.R.; Kleinberger, T.; Akusjarvi, G. The adenovirus E4-ORF4 splicing enhancer protein interacts with a subset of phosphorylated SR proteins. EMBO J. 2001, 20, 864–871. [Google Scholar]
- Muller, U.; Kleinberger, T.; Shenk, T. Adenovirus E4orf4 protein reduces phosphorylation of c-Fos and E1A proteins while simultaneously reducing the level of AP-1. J. Virol. 1992, 66, 5867–5878. [Google Scholar] [PubMed]
- Kleinberger, T.; Shenk, T. Adenovirus E4orf4 protein binds to protein phosphatase 2A, and the complex down regulates E1A-enhanced junB transcription. J. Virol. 1993, 67, 7556–7560. [Google Scholar] [PubMed]
- Bondesson, M.; Ohman, K.; Manervik, M.; Fan, S.; Akusjarvi, G. Adenovirus E4 open reading frame 4 protein autoregulates E4 transcription by inhibiting E1A transactivation of the E4 promoter. J. Virol. 1996, 70, 3844–3851. [Google Scholar] [PubMed]
- Lu, Y.; Kucharski, T.J.; Gamache, I.; Blanchette, P.; Branton, P.E.; Teodoro, J.G. Interaction of adenovirus type 5 E4orf4 with the nuclear pore subunit Nup205 is required for proper viral gene expression. J. Virol. 2014, 88, 13249–13259. [Google Scholar] [CrossRef] [PubMed]
- Muhlemann, O.; Yue, B.G.; Petersen-Mahrt, S.; Akusjarvi, G. A novel type of splicing enhancer regulating adenovirus pre-mRNA splicing. Mol. Cell. Biol. 2000, 20, 2317–2325. [Google Scholar] [CrossRef] [PubMed]
- Lutzelberger, M.; Backstrom, E.; Akusjarvi, G. Substrate-dependent differences in U2AF requirement for splicing in adenovirus-infected cell extracts. J. Biol. Chem. 2005, 280, 25478–25484. [Google Scholar] [CrossRef] [PubMed]
- Muhlemann, O.; Kreivi, J.P.; Akusjarvi, G. Enhanced splicing of nonconsensus 3' splice sites late during adenovirus infection. J. Virol. 1995, 69, 7324–7327. [Google Scholar] [PubMed]
- Russell, W.C. Adenoviruses: Update on structure and function. J. Gen. Virol. 2009, 90, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Cepko, C.L.; Sharp, P.A. Assembly of adenovirus major capsid protein is mediated by a nonvirion protein. Cell 1982, 31, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Xi, Q.; Cuesta, R.; Schneider, R.J. Tethering of eIF4G to adenoviral mRNAs by viral 100k protein drives ribosome shunting. Genes Dev. 2004, 18, 1997–2009. [Google Scholar] [CrossRef] [PubMed]
- Ostapchuk, P.; Anderson, M.E.; Chandrasekhar, S.; Hearing, P. The L4 22-kilodalton protein plays a role in packaging of the adenovirus genome. J. Virol. 2006, 80, 6973–6981. [Google Scholar] [CrossRef] [PubMed]
- Ahi, Y.S.; Vemula, S.V.; Mittal, S.K. Adenoviral E2 IVa2 protein interacts with L4 33K protein and E2 DNA-binding protein. J. Gen. Virol. 2013, 94, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.; Leppard, K.N. The human adenovirus 5 L4 promoter is activated by cellular stress response protein p53. J. Virol. 2013, 87, 11617–11625. [Google Scholar] [CrossRef] [PubMed]
- Backstrom, E.; Kaufmann, K.B.; Lan, X.; Akusjarvi, G. Adenovirus L4–22 k stimulates major late transcription by a mechanism requiring the intragenic late-specific transcription factor-binding site. Virus Res. 2010, 151, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Tribouley, C.; Lutz, P.; Staub, A.; Kedinger, C. The product of the adenovirus intermediate gene IVa2 is a transcriptional activator of the major late promoter. J. Virol. 1994, 68, 4450–4457. [Google Scholar] [PubMed]
- Tormanen, H.; Backstrom, E.; Carlsson, A.; Akusjarvi, G. L4–33k, an adenovirus-encoded alternative RNA splicing factor. J. Biol. Chem. 2006, 281, 36510–36517. [Google Scholar] [CrossRef] [PubMed]
- Ewing, S.G.; Byrd, S.A.; Christensen, J.B.; Tyler, R.E.; Imperiale, M.J. Ternary complex formation on the adenovirus packaging sequence by the IVa2 and L4 22-kilodalton proteins. J. Virol. 2007, 81, 12450–12457. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Orozco, D.; Hearing, P. The adenovirus L4–22 k protein is multifunctional and is an integral component of crucial aspects of infection. J. Virol. 2012, 86, 10474–10483. [Google Scholar] [CrossRef] [PubMed]
- Guimet, D.; Hearing, P. The adenovirus L4–22k protein has distinct functions in the posttranscriptional regulation of gene expression and encapsidation of the viral genome. J. Virol. 2013, 87, 7688–7699. [Google Scholar] [CrossRef] [PubMed]
- Yachdav, G.; Kloppmann, E.; Kajan, L.; Hecht, M.; Goldberg, T.; Hamp, T.; Honigschmid, P.; Schafferhans, A.; Roos, M.; Bernhofer, M.; et al. Predictprotein—An open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 2014, 42, W337–W343. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.; Lee, W.; Berk, A.J. High-level transcription from the adenovirus major late promoter requires downstream binding sites for late-phase-specific factors. J. Virol. 1990, 64, 51–60. [Google Scholar] [PubMed]
- Lutz, P.; Kedinger, C. Properties of the adenovirus IVa2 gene product, an effector of late-phase-dependent activation of the major late promoter. J. Virol. 1996, 70, 1396–1405. [Google Scholar] [PubMed]
- Jansen-Durr, P.; Mondesert, G.; Kedinger, C. Replication-dependent activation of the adenovirus major late promoter is mediated by the increased binding of a transcription factor to sequences in the first intron. J. Virol. 1989, 63, 5124–5132. [Google Scholar] [PubMed]
- Mondesert, G.; Tribouley, C.; Kedinger, C. Identification of a novel downstream binding protein implicated in late-phase-specific activation of the adenovirus major late promotor. Nucleic Acids Res. 1992, 20, 3881–3889. [Google Scholar] [CrossRef] [PubMed]
- Ostapchuk, P.; Hearing, P. Control of adenovirus packaging. J. Cell. Biochem. 2005, 96, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.; LeRoy, G.; Bridge, G.; Flint, S.J. The adenovirus L4 33-kilodalton protein binds to intragenic sequences of the major late promoter required for late phase-specific stimulation of transcription. J. Virol. 2007, 81, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Tollefson, A.E.; Scaria, A.; Saha, S.K.; Wold, W.S. The 11,600-MW protein encoded by region E3 of adenovirus is expressed early but is greatly amplified at late stages of infection. J. Virol. 1992, 66, 3633–3642. [Google Scholar] [PubMed]
- Farley, D.C.; Brown, J.L.; Leppard, K.N. Activation of the early-late switch in adenovirus type 5 major late transcription unit expression by l4 gene products. J. Virol. 2004, 78, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Guimet, D.; Hearing, P. The adenovirus L4–33k protein regulates both late gene expression patterns and viral DNA packaging. J. Virol. 2013, 87, 6739–6747. [Google Scholar] [CrossRef] [PubMed]
- Fessler, S.P.; Young, C.S. The role of the L4 33k gene in adenovirus infection. Virology 1999, 263, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Finnen, R.L.; Biddle, J.F.; Flint, J. Truncation of the human adenovirus type 5 L4 33-kDa protein: Evidence for an essential role of the carboxy-terminus in the viral infectious cycle. Virology 2001, 289, 388–399. [Google Scholar] [CrossRef]
- Kulshreshtha, V.; Babiuk, L.A.; Tikoo, S.K. Role of bovine adenovirus-3 33k protein in viral replication. Virology 2004, 323, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Ostberg, S.; Tormanen Persson, H.; Akusjarvi, G. Serine 192 in the tiny rs repeat of the adenoviral L4–33k splicing enhancer protein is essential for function and reorganization of the protein to the periphery of viral replication centers. Virology 2012, 433, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, C.F.; Lejeune, F.; Stevenin, J. Broad specificity of SR (serine/arginine) proteins in the regulation of alternative splicing of pre-messenger RNA. Prog. Nucleic Acid Res. Mol. Biol. 2004, 78, 37–88. [Google Scholar] [PubMed]
- Shepard, P.J.; Hertel, K.J. The SR protein family. Genome Biol. 2009, 10, 242. [Google Scholar] [CrossRef] [PubMed]
- Manley, J.L.; Krainer, A.R. A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins). Genes Dev. 2010, 24, 1073–1074. [Google Scholar] [CrossRef] [PubMed]
- Kulshreshtha, V.; Ayalew, L.E.; Islam, A.; Tikoo, S.K. Conserved arginines of bovine adenovirus-3 33k protein are important for transportin-3 mediated transport and virus replication. PLoS One 2014, 9, e101216. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.C.; Lin, R.I.; Tarn, W.Y. Transportin-SR2 mediates nuclear import of phosphorylated SR proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 10154–10159. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.; Church, G.M. Collection and motif-based prediction of phosphorylation sites in human viruses. Sci. Signal. 2010, 3, rs2. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, A.S.; Ponticelli, A.; Berk, A.J.; Gaynor, R.B. Genetic mapping of a major site of phosphorylation in adenovirus type 2 E1A proteins. J. Virol. 1986, 59, 14–22. [Google Scholar] [PubMed]
- Tremblay, M.L.; McGlade, C.J.; Gerber, G.E.; Branton, P.E. Identification of the phosphorylation sites in early region 1A proteins of adenovirus type 5 by amino acid sequencing of peptide fragments. J. Biol. Chem. 1988, 263, 6375–6383. [Google Scholar] [PubMed]
- Smith, C.L.; Debouck, C.; Rosenberg, M.; Culp, J.S. Phosphorylation of serine residue 89 of human adenovirus e1a proteins is responsible for their characteristic electrophoretic mobility shifts, and its mutation affects biological function. J. Virol. 1989, 63, 1569–1577. [Google Scholar] [PubMed]
- Whalen, S.G.; Marcellus, R.C.; Barbeau, D.; Branton, P.E. Importance of the ser-132 phosphorylation site in cell transformation and apoptosis induced by the adenovirus type 5 E1A protein. J. Virol. 1996, 70, 5373–5383. [Google Scholar] [PubMed]
- Whalen, S.G.; Marcellus, R.C.; Whalen, A.; Ahn, N.G.; Ricciardi, R.P.; Branton, P.E. Phosphorylation within the transactivation domain of adenovirus E1A protein by mitogen-activated protein kinase regulates expression of early region 4. J. Virol. 1997, 71, 3545–3553. [Google Scholar] [PubMed]
- Turnell, A.S.; Grand, R.J.; Gorbea, C.; Zhang, X.; Wang, W.; Mymryk, J.S.; Gallimore, P.H. Regulation of the 26s proteasome by adenovirus E1A. EMBO J. 2000, 19, 4759–4773. [Google Scholar] [CrossRef] [PubMed]
- Dumont, D.J.; Marcellus, R.C.; Bayley, S.T.; Branton, P.E. Role of phosphorylation near the amino terminus of adenovirus type 5 early region 1a proteins. J. Gen. Virol. 1993, 74, 583–595. [Google Scholar] [CrossRef] [PubMed]
- McGlade, C.J.; Tremblay, M.L.; Branton, P.E. Mapping of a phosphorylation site in the 176R (19 kDa) early region 1B protein of human adenovirus type 5. Virology 1989, 168, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Takayesu, D.; Teodoro, J.G.; Whalen, S.G.; Branton, P.E. Characterization of the 55k adenovirus type 5 E1B product and related proteins. J. Gen. Virol. 1994, 75, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Cleghon, V.; Piderit, A.; Brough, D.E.; Klessig, D.F. Phosphorylation of the adenovirus DNA-binding protein and epitope mapping of monoclonal antibodies against it. Virology 1993, 197, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, N. Phosphoproteins of adenovirus 2. Virology 1978, 87, 366–383. [Google Scholar] [CrossRef] [PubMed]
- Gambke, C.; Deppert, W. Late nonstructural 100,000- and 33,000-dalton proteins of adenovirus type 2. I. Subcellular localization during the course of infection. J. Virol. 1981, 40, 585–593. [Google Scholar] [PubMed]
- Lichtenstein, D.L.; Krajcsi, P.; Esteban, D.J.; Tollefson, A.E.; Wold, W.S. Adenovirus ridbeta subunit contains a tyrosine residue that is critical for RID-mediated receptor internalization and inhibition of fas- and trail-induced apoptosis. J. Virol. 2002, 76, 11329–11342. [Google Scholar] [CrossRef] [PubMed]
- Gingras, M.C.; Champagne, C.; Roy, M.; Lavoie, J.N. Cytoplasmic death signal triggered by src-mediated phosphorylation of the adenovirus E4ORF4 protein. Mol. Cell. Biol. 2002, 22, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.C.; Blair, G.E. Polypeptide phosphorylation in adenovirus-infected cells. J. Gen. Virol. 1977, 34, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra, M.; Nakano, R.; Mohan, P.M.; Rawitch, A.B.; Padmanabhan, R. Adenovirus DNA polymerase is a phosphoprotein. J. Biol. Chem. 1993, 268, 442–448. [Google Scholar] [PubMed]
- Bergstrom Lind, S.; Artemenko, K.A.; Elfineh, L.; Zhao, Y.; Bergquist, J.; Pettersson, U. The phosphoproteome of the adenovirus type 2 virion. Virology 2012, 433, 253–261. [Google Scholar]
- Bergstrom Lind, S.; Artemenko, K.A.; Elfineh, L.; Zhao, Y.; Bergquist, J.; Pettersson, U. Post translational modifications in adenovirus type 2. Virology 2013, 447, 104–111. [Google Scholar]
- Tormanen Persson, H.; Aksaas, A.K.; Kvissel, A.K.; Punga, T.; Engstrom, A.; Skalhegg, B.S.; Akusjarvi, G. Two cellular protein kinases, DNA-PK and PKA, phosphorylate the adenoviral L4–33k protein and have opposite effects on L1 alternative rna splicing. PLoS One 2012, 7, e31871. [Google Scholar]
- Turnell, A.S.; Grand, R.J. DNA viruses and the cellular DNA-damage response. J. Gen. Virol. 2012, 93, 2076–2097. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.; Rohleder, K.; Ketner, G. Adenovirus E4 34k and E4 11k inhibit double strand break repair and are physically associated with the cellular DNA-dependent protein kinase. Virology 1999, 263, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, B.P.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Weiden, M.D.; Ginsberg, H.S. Deletion of the E4 region of the genome produces adenovirus DNA concatemers. Proc. Natl. Acad. Sci. USA 1994, 91, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Ornelles, D.A. Inactivating intracellular antiviral responses during adenovirus infection. Oncogene 2005, 24, 7686–7696. [Google Scholar] [CrossRef] [PubMed]
- Ohman, K.; Nordqvist, K.; Akusjarvi, G. Two adenovirus proteins with redundant activities in virus growth facilitates tripartite leader mrna accumulation. Virology 1993, 194, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Nordqvist, K.; Ohman, K.; Akusjarvi, G. Human adenovirus encodes two proteins which have opposite effects on accumulation of alternatively spliced mRNAs. Mol. Cell. Biol. 1994, 14, 437–445. [Google Scholar] [PubMed]
- Zhou, Z.; Fu, X.D. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [PubMed]
- Blom, N.; Sicheritz-Ponten, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef] [PubMed]
- Dhurandhar, N.V. A framework for identification of infections that contribute to human obesity. Lancet Infect. Dis. 2011, 11, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Ghebremedhin, B. Human adenovirus: Viral pathogen with increasing importance. Eur. J. Microbiol. Immunol. 2014, 4, 26–33. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biasiotto, R.; Akusjärvi, G. Regulation of Human Adenovirus Alternative RNA Splicing by the Adenoviral L4-33K and L4-22K Proteins. Int. J. Mol. Sci. 2015, 16, 2893-2912. https://doi.org/10.3390/ijms16022893
Biasiotto R, Akusjärvi G. Regulation of Human Adenovirus Alternative RNA Splicing by the Adenoviral L4-33K and L4-22K Proteins. International Journal of Molecular Sciences. 2015; 16(2):2893-2912. https://doi.org/10.3390/ijms16022893
Chicago/Turabian StyleBiasiotto, Roberta, and Göran Akusjärvi. 2015. "Regulation of Human Adenovirus Alternative RNA Splicing by the Adenoviral L4-33K and L4-22K Proteins" International Journal of Molecular Sciences 16, no. 2: 2893-2912. https://doi.org/10.3390/ijms16022893
APA StyleBiasiotto, R., & Akusjärvi, G. (2015). Regulation of Human Adenovirus Alternative RNA Splicing by the Adenoviral L4-33K and L4-22K Proteins. International Journal of Molecular Sciences, 16(2), 2893-2912. https://doi.org/10.3390/ijms16022893