
PEDF Improves Cardiac Function in Rats with Acute Myocardial Infarction via Inhibiting Vascular Permeability and Cardiomyocyte Apoptosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
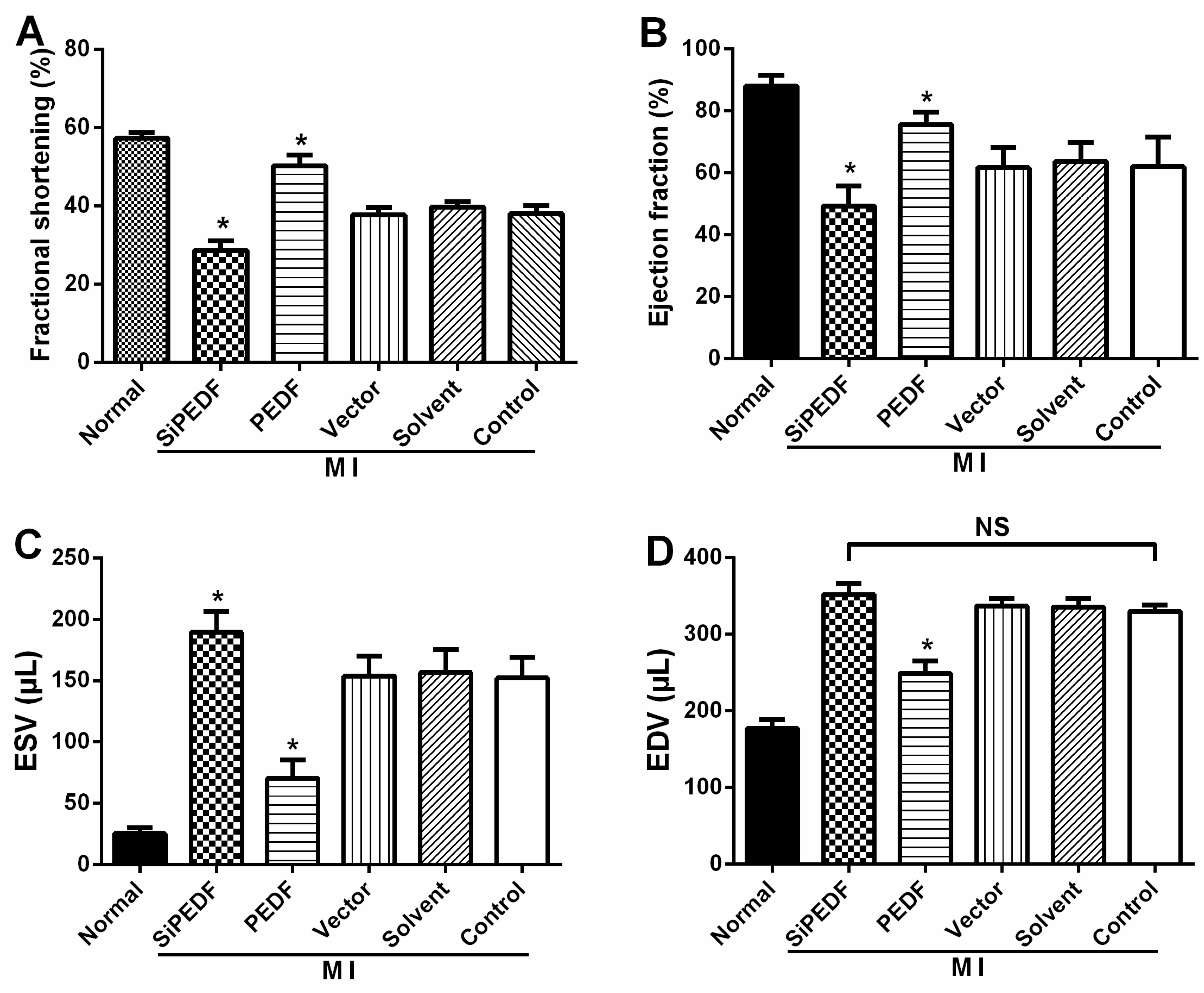
2.1. PEDF Improves Cardiac Function


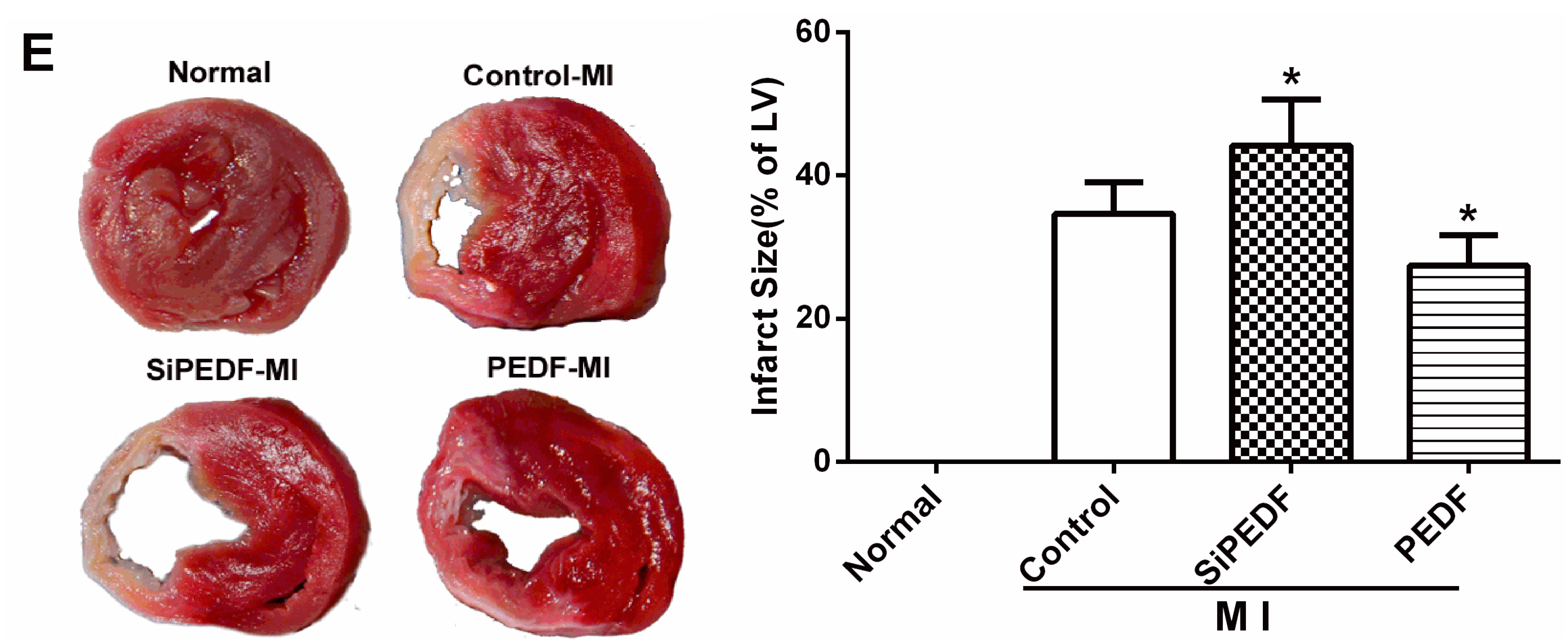
2.2. PEDF Reduces Myocardial Infarct Size
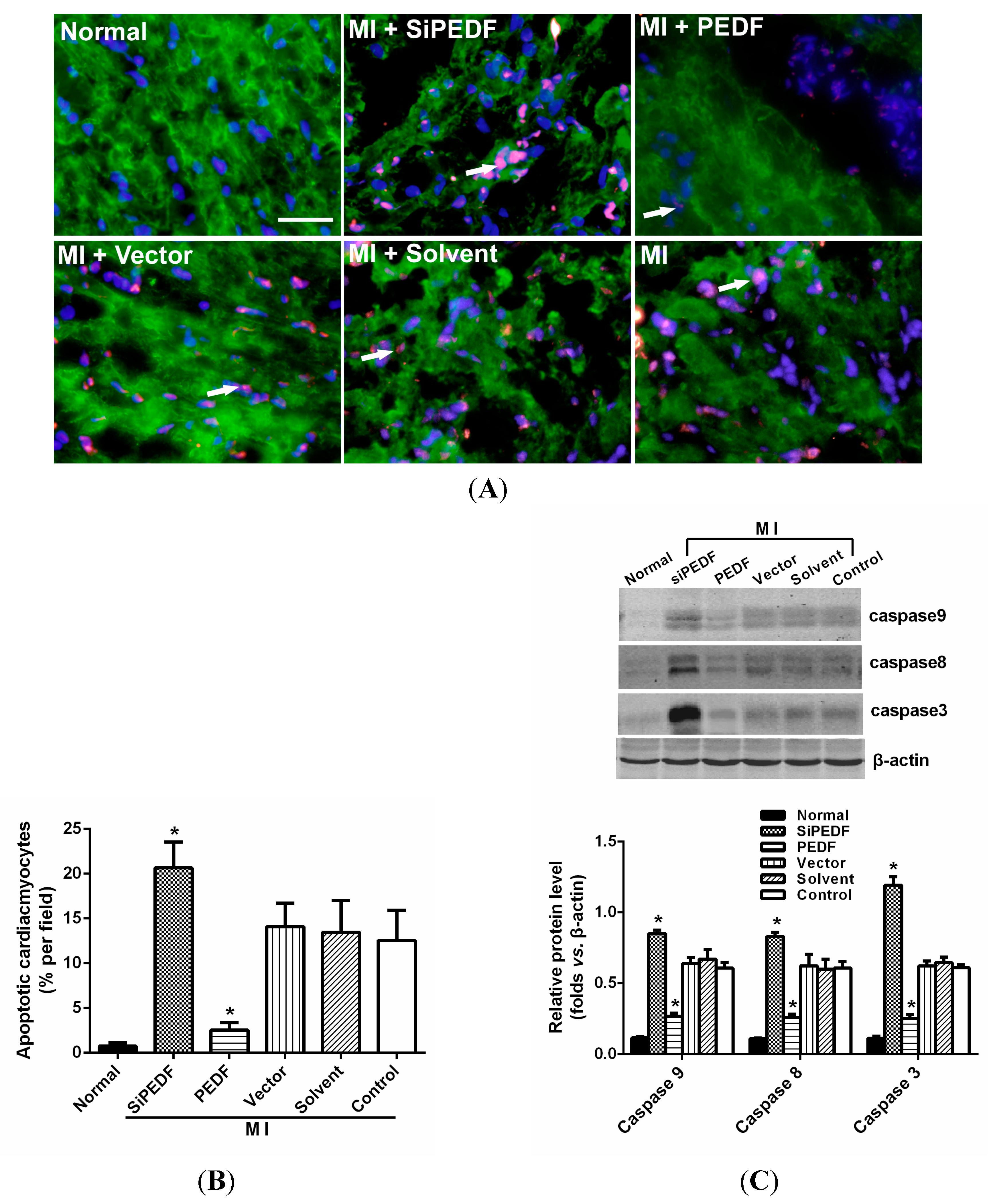
2.3. PEDF Reduces Cardiomyocyte Apoptosis in the Ischemic Myocardium

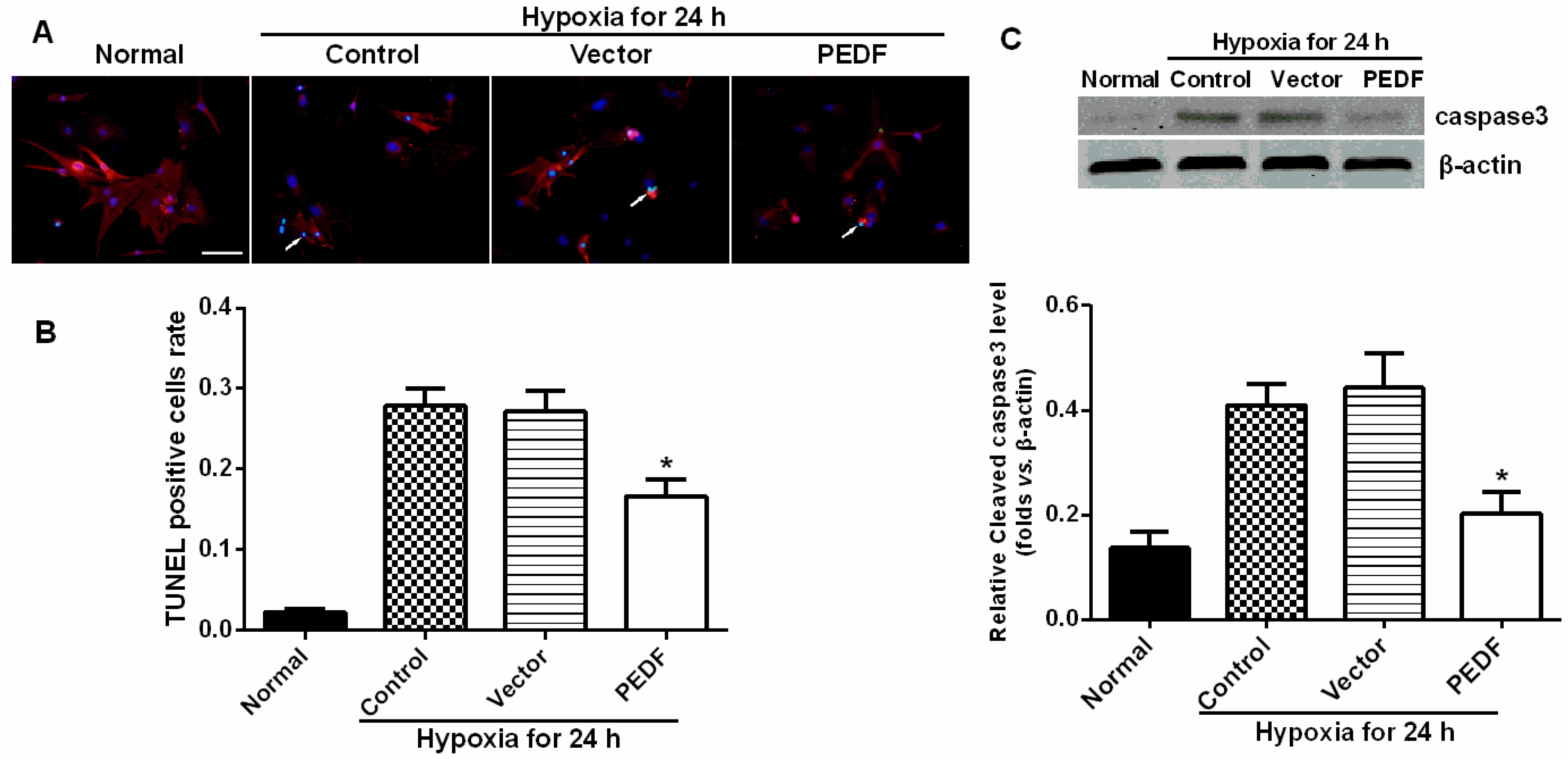
2.4. PEDF Protects against Hypoxia-Induced Cardiomyocyte Apoptosis

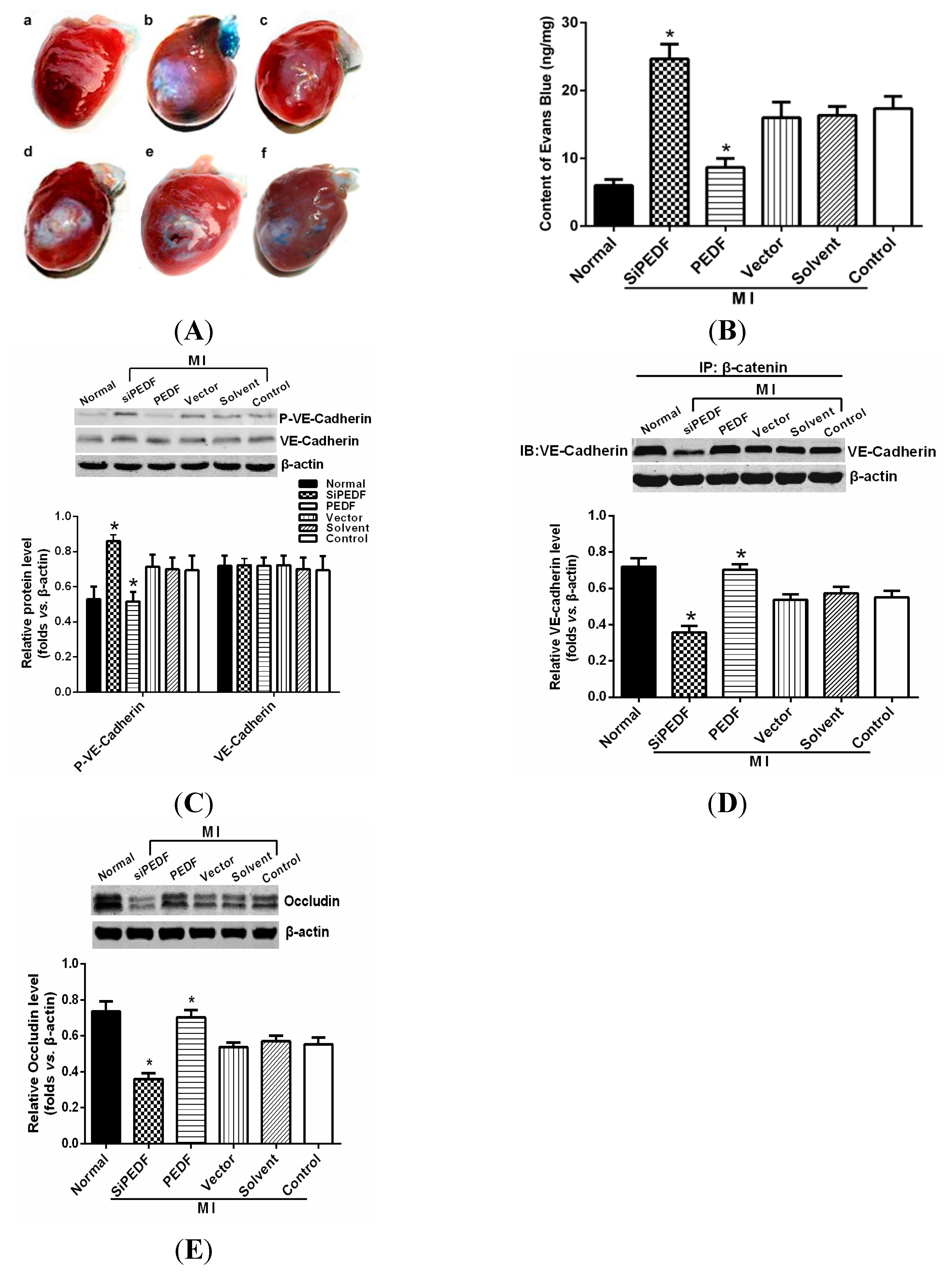
2.5. PEDF Prevents Vascular Leakage

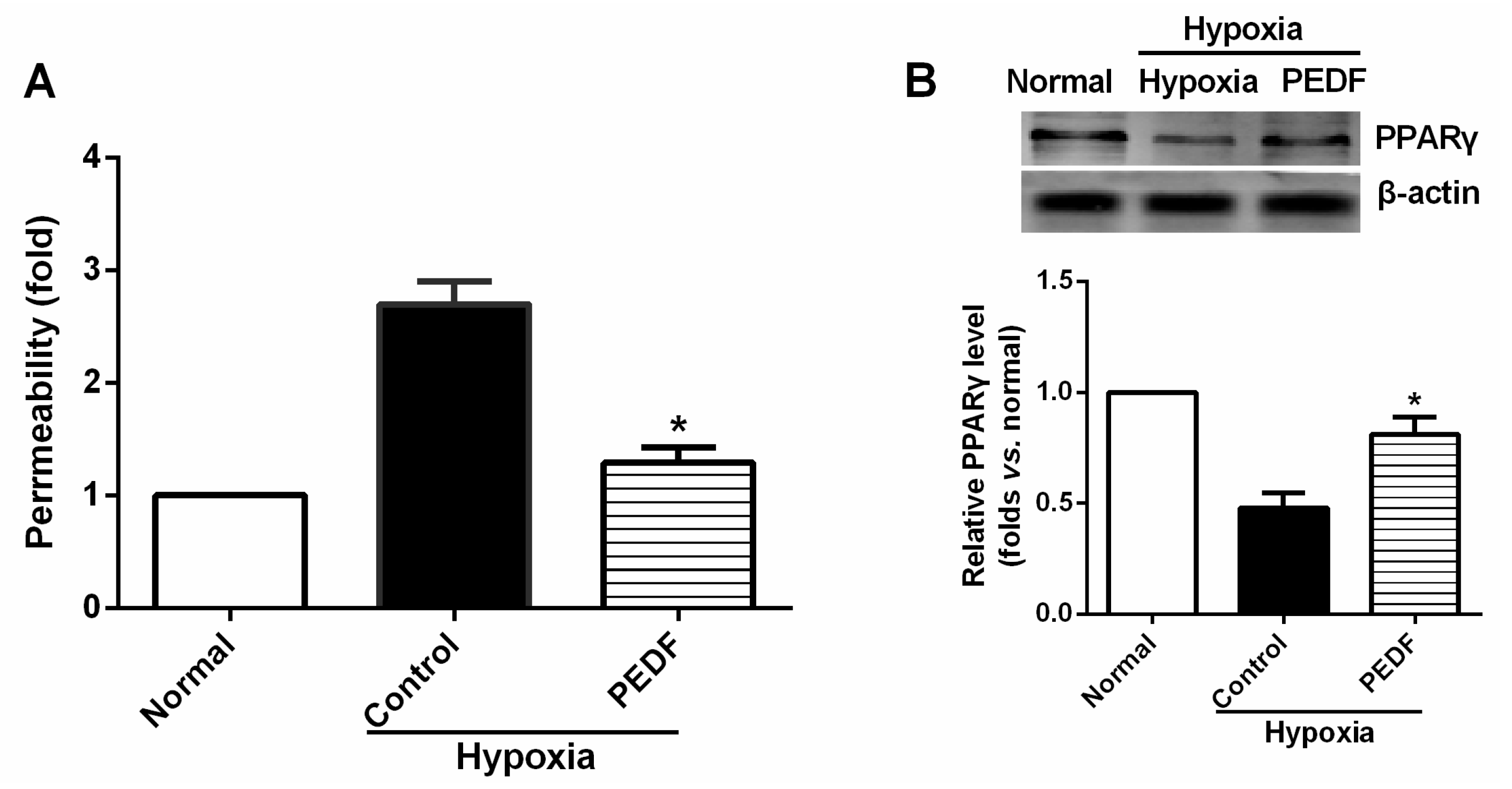
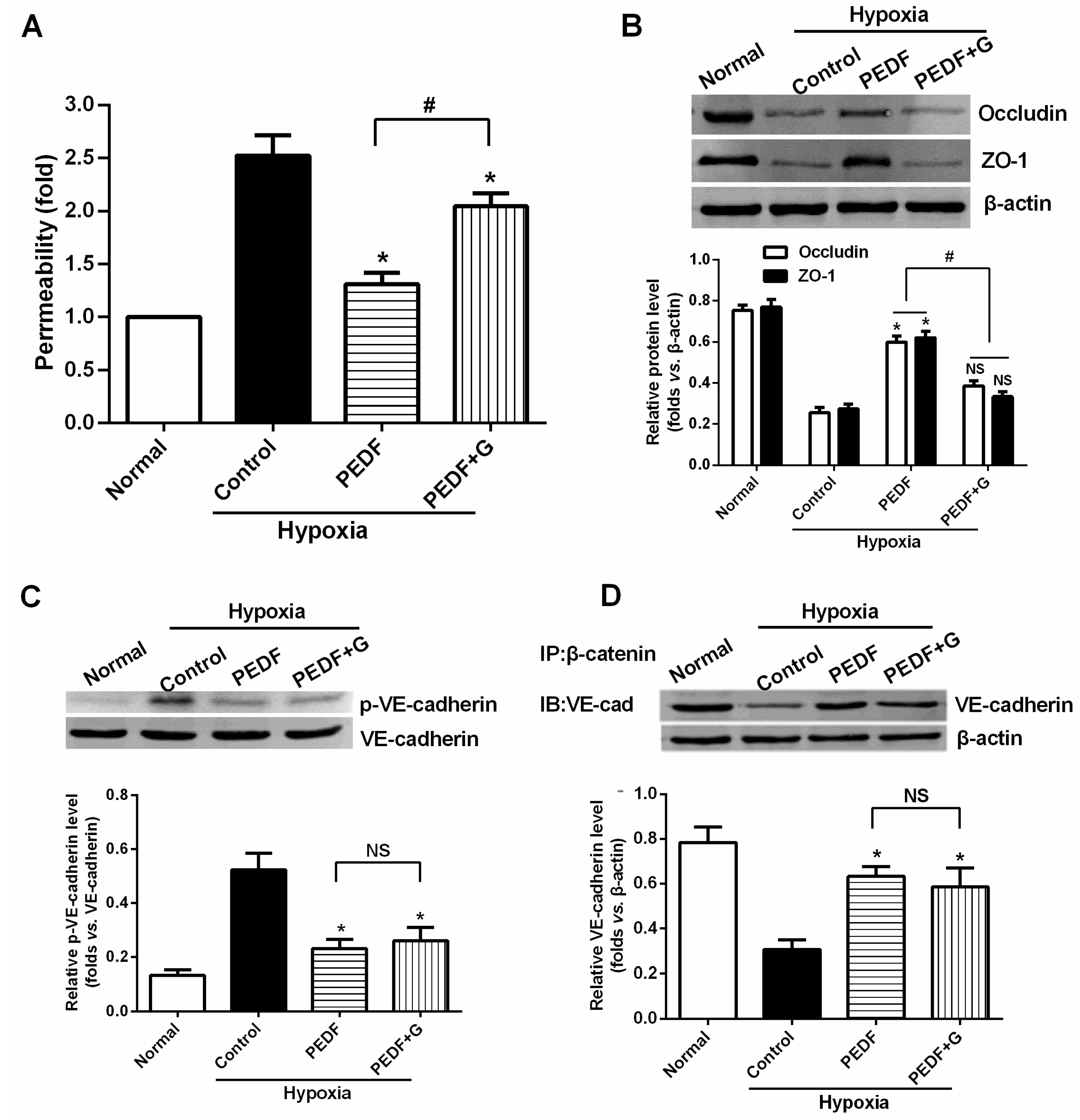
2.6. PEDF Prevents Hypoxia-Induced Rat Aortic Endothelial Cell Permeability

2.7. PEDF Inhibits Hypoxia-Induced RAECs Permeability through PPARγ-Dependent TJ Production

3. Discussion
4. Experimental Section
4.1. Recombinant Lentivirus Constructs and Viral Production
4.2. Animal Model and Intramyocardial Gene Delivery
4.3. Animal Cardiac Function Evaluation
4.4. Quantification of Myocardial Infarct Size
4.5. Cardiomyocyte Apoptosis Assay in Vivo
4.6. Vascular Permeability Measurement
4.7. Western Blot Analysis and Co-Immunoprecipitation
4.8. Preparations of PEDF Protein
4.9. Cardiomyocyte Culture and Cardiomyocyte Apoptosis Assay in Vitro
4.10. RAECs Culture and Permeability Measurements in Vitro
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lloyd-Jones, D.; Adams, R.; Carnethon, M.; de Simone, G.; Ferguson, T.B.; Flegal, K.; Ford, E.; Furie, K.; Go, A.; Greenlund, K.; et al. Heart disease and stroke statistics—2009 Update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2009, 119, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.; Whelan, R.S.; Kitsis, R.N. Mechanisms of cell death in heart disease. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Filleur, S.; Nelius, T.; de Riese, W.; Kennedy, R.C. Characterization of PEDF: A multi-functional serpin family protein. J. Cell. Biochem. 2009, 106, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Drevon, C.A. Fatty acids and expression of adipokines. Biochim. Biophys. Acta 2005, 1740, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Sawant, S.; Aparicio, S.; Tink, A.R.; Lara, N.; Barnstable, C.J.; Tombran-Tink, J. Regulation of factors controlling angiogenesis in liver development: A role for PEDF in the formation and maintenance of normal vasculature. Biochem. Biophys. Res. Commun. 2004, 325, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T.; Nakamura, K. Atheroprotective properties of pigment epithelium-derived factor (PEDF) in cardiometabolic disorders. Curr. Pharm. Des. 2009, 15, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.X.; Wang, J.J.; Gao, G.; Shao, C.; Mott, R.; Ma, J.X. Pigment epithelium-derived factor (PEDF) is an endogenous antiinflammatory factor. FASEB J. 2006, 20, 323–325. [Google Scholar] [PubMed]
- Truong, A.; Wong, T.Y.; Khachigian, L.M. Emerging therapeutic approaches in the management of retinal angiogenesis and edema. J. Mol. Med. 2011, 89, 343–361. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, N.I.; Volpert, O.V.; Jimenez, B. Pigment epithelium-derived factor as a multifunctional antitumor factor. J. Mol. Med. 2007, 85, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Wu, L.; Qi, X.; Li Calzi, S.; Caballero, S.; Shaw, L.; Ruan, Q.; Grant, M.B.; Boulton, M.E. PEDF regulates vascular permeability by a γ-secretase-mediated pathway. PLoS One 2011, 6, e21164. [Google Scholar] [CrossRef] [PubMed]
- Mejias, M.; Coch, L.; Berzigotti, A.; Garcia-Pras, E.; Gallego, J.; Bosch, J.; Fernandez, M. Antiangiogenic and antifibrogenic activity of pigment epithelium-derived factor (PEDF) in bile duct-ligated portal hypertensive rats. Gut 2014, 64, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ma, X.; Zhou, M.; Pan, X.; Ni, J.; Gao, M.; Lu, Z.; Hang, J.; Bao, Y.; Jia, W. Serum pigment epithelium-derived factor levels are independently correlated with the presence of coronary artery disease. Cardiovasc. Diabetol. 2013, 12. [Google Scholar] [CrossRef]
- Ueda, S.; Yamagishi, S.; Matsui, T.; Jinnouchi, Y.; Imaizumi, T. Administration of pigment epithelium-derived factor inhibits left ventricular remodeling and improves cardiac function in rats with acute myocardial infarction. Am. J. Pathol. 2011, 178, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Li, J.K.; Liang, H.L.; Li, Z.; Gu, C.H.; Yi, D.H.; Pei, J.M. Pigment epithelium-derived factor promotes Fas-induced cardiomyocyte apoptosis via its receptor phospholipase A2. Life Sci. 2014, 99, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Rychli, K.; Niessner, A.; Hohensinner, P.J.; Mahdy Ali, K.; Kaun, C.; Neuhold, S.; Zorn, G.; Richter, B.; Hulsmann, M.; Berger, R.; et al. Prognostic value of pigment epithelium-derived factor in patients with advanced heart failure. Chest 2010, 138, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.J.; Fan, Y.; Hamblin, M.; Zhang, J.; Zhu, T.; Li, S.; Hawse, J.R.; Subramaniam, M.; Song, C.Z.; Urrutia, R.; et al. KLF11 mediates PPARγ cerebrovascular protection in ischaemic stroke. Brain 2013, 136, 1274–1287. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, N.; Kojima, T.; Go, M.; Ohkuni, T.; Koizumi, J.; Kamekura, R.; Masaki, T.; Murata, M.; Tanaka, S.; Fuchimoto, J.; et al. PPARγ agonists upregulate the barrier function of tight junctions via a PKC pathway in human nasal epithelial cells. Pharmacol. Res. 2010, 61, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.; Yang, Y.C.; Chen, S.L.; Kuo, P.C.; Sytwu, H.K.; Cheng, H.C.; Tsao, Y.P. Pigment epithelium-derived factor induces THP-1 macrophage apoptosis and necrosis by the induction of the peroxisome proliferator-activated receptor γ. Mol. Immunol. 2008, 45, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.J.; Kim, P.; Lu, Y.F.; Feingold, K.R. PPARgamma activators stimulate aquaporin 3 expression in keratinocytes/epidermis. Exp. Dermatol. 2011, 20, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Whelan, R.S.; Kaplinskiy, V.; Kitsis, R.N. Cell death in the pathogenesis of heart disease: Mechanisms and significance. Annu. Rev. Physiol. 2010, 72, 19–44. [Google Scholar] [CrossRef] [PubMed]
- Wencker, D.; Chandra, M.; Nguyen, K.; Miao, W.; Garantziotis, S.; Factor, S.M.; Shirani, J.; Armstrong, R.C.; Kitsis, R.N. A mechanistic role for cardiac myocyte apoptosis in heart failure. J. Clin. Investig. 2003, 111, 1497–1504. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, P.; Locatelli-Hoops, S.; Kenealey, J.; DesJardin, J.; Notari, L.; Becerra, S.P. Pigment epithelium-derived factor (PEDF) prevents retinal cell death via PEDF Receptor (PEDF-R): Identification of a functional ligand binding site. J. Biol. Chem. 2013, 288, 23928–23942. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, H.; Zhuang, W.; Yuan, G.; Sun, T.; Jiang, X.; Zhou, Z.; Yuan, H.; Zhang, Z.; Dong, H. PEDF and PEDF-derived peptide 44mer protect cardiomyocytes against hypoxia-induced apoptosis and necroptosis via anti-oxidative effect. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Maczewski, M.; Mackiewicz, U. Effect of metoprolol and ivabradine on left ventricular remodelling and Ca2+ handling in the post-infarction rat heart. Cardiovasc. Res. 2008, 79, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Ahmet, I.; Lakatta, E.G.; Talan, M.I. Pharmacological stimulation of β2-adrenergic receptors (β2AR) enhances therapeutic effectiveness of β1AR blockade in rodent dilated ischemic cardiomyopathy. Heart Fail. Rev. 2005, 10, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.; Zhang, L.; Duan, L.; Wang, X.; Min, Y.; Yu, H. Extracellular vesicles derived from human bone marrow mesenchymal stem cells promote angiogenesis in a rat myocardial infarction model. J. Mol. Med. 2014, 92, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yuan, Y.L.; Wang, Z.; Jiang, B.; Zhang, C.S.; Wang, Q.; Xu, X.H.; Dong, H.Y.; Zhang, Z.M. Sequential, timely and controlled expression of hVEGF165 and Ang-1 effectively improves functional angiogenesis and cardiac function in vivo. Gene Ther. 2013, 20, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Rutanen, J.; Rissanen, T.T.; Markkanen, J.E.; Gruchala, M.; Silvennoinen, P.; Kivela, A.; Hedman, A.; Hedman, M.; Heikura, T.; Orden, M.R.; et al. Adenoviral catheter-mediated intramyocardial gene transfer using the mature form of vascular endothelial growth factor-D induces transmural angiogenesis in porcine heart. Circulation 2004, 109, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Y.; Fang, J.; Zhang, H. FIZZ1 could enhance the angiogenic ability of rat aortic endothelial cells. Int. J. Clin. Exp. Pathol. 2013, 6, 1847–1853. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Wang, Z.; Feng, S.-J.; Xu, L.; Shi, H.-X.; Chen, L.-L.; Yuan, G.-D.; Yan, W.; Zhuang, W.; Zhang, Y.-Q.; et al. PEDF Improves Cardiac Function in Rats with Acute Myocardial Infarction via Inhibiting Vascular Permeability and Cardiomyocyte Apoptosis. Int. J. Mol. Sci. 2015, 16, 5618-5634. https://doi.org/10.3390/ijms16035618
Zhang H, Wang Z, Feng S-J, Xu L, Shi H-X, Chen L-L, Yuan G-D, Yan W, Zhuang W, Zhang Y-Q, et al. PEDF Improves Cardiac Function in Rats with Acute Myocardial Infarction via Inhibiting Vascular Permeability and Cardiomyocyte Apoptosis. International Journal of Molecular Sciences. 2015; 16(3):5618-5634. https://doi.org/10.3390/ijms16035618
Chicago/Turabian StyleZhang, Hao, Zheng Wang, Shou-Jie Feng, Lei Xu, He-Xian Shi, Li-Li Chen, Guang-Da Yuan, Wei Yan, Wei Zhuang, Yi-Qian Zhang, and et al. 2015. "PEDF Improves Cardiac Function in Rats with Acute Myocardial Infarction via Inhibiting Vascular Permeability and Cardiomyocyte Apoptosis" International Journal of Molecular Sciences 16, no. 3: 5618-5634. https://doi.org/10.3390/ijms16035618
APA StyleZhang, H., Wang, Z., Feng, S. -J., Xu, L., Shi, H. -X., Chen, L. -L., Yuan, G. -D., Yan, W., Zhuang, W., Zhang, Y. -Q., Zhang, Z. -M., & Dong, H. -Y. (2015). PEDF Improves Cardiac Function in Rats with Acute Myocardial Infarction via Inhibiting Vascular Permeability and Cardiomyocyte Apoptosis. International Journal of Molecular Sciences, 16(3), 5618-5634. https://doi.org/10.3390/ijms16035618