
RNA-Seq Transcriptome Analysis of Maize Inbred Carrying Nicosulfuron-Tolerant and Nicosulfuron-Susceptible Alleles


Abstract
:1. Introduction
2. Results and Discussion
2.1. Phenotypic Responses to Nicosulfuron
2.2. RNA-Seq Analysis Aligned with the Maize Reference Genome Sequence

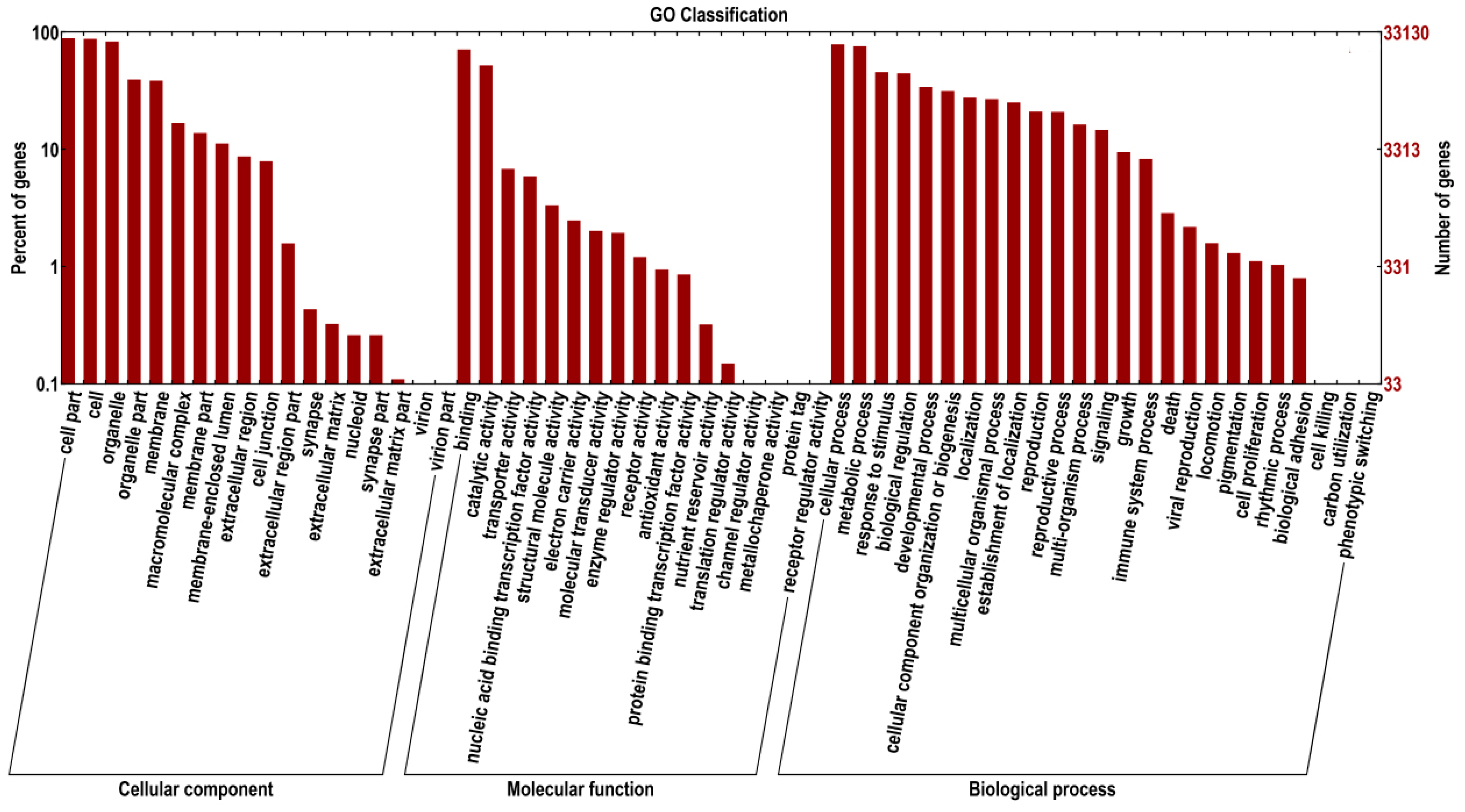
2.3. Gene Functional Annotation by GO, and KEGG

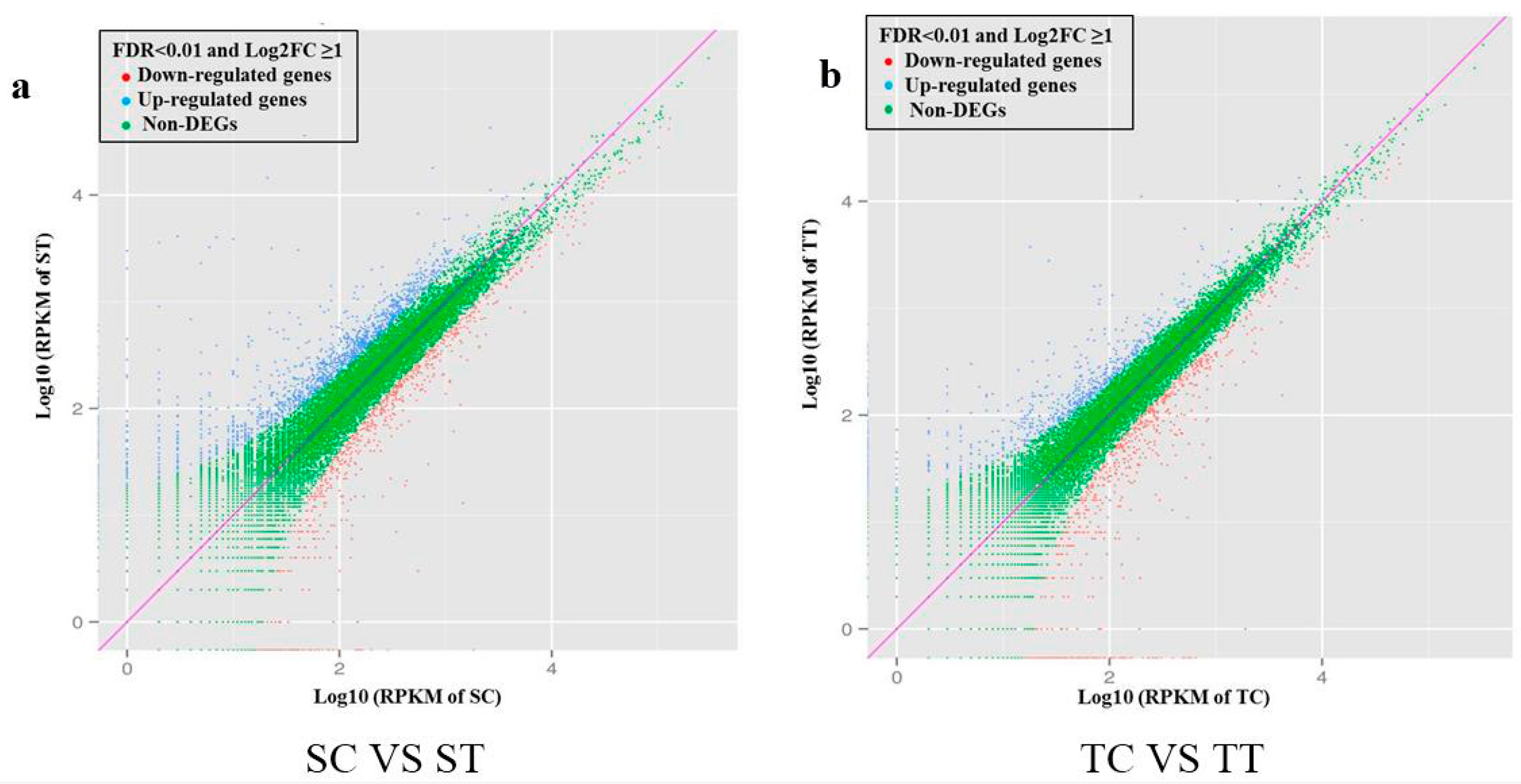
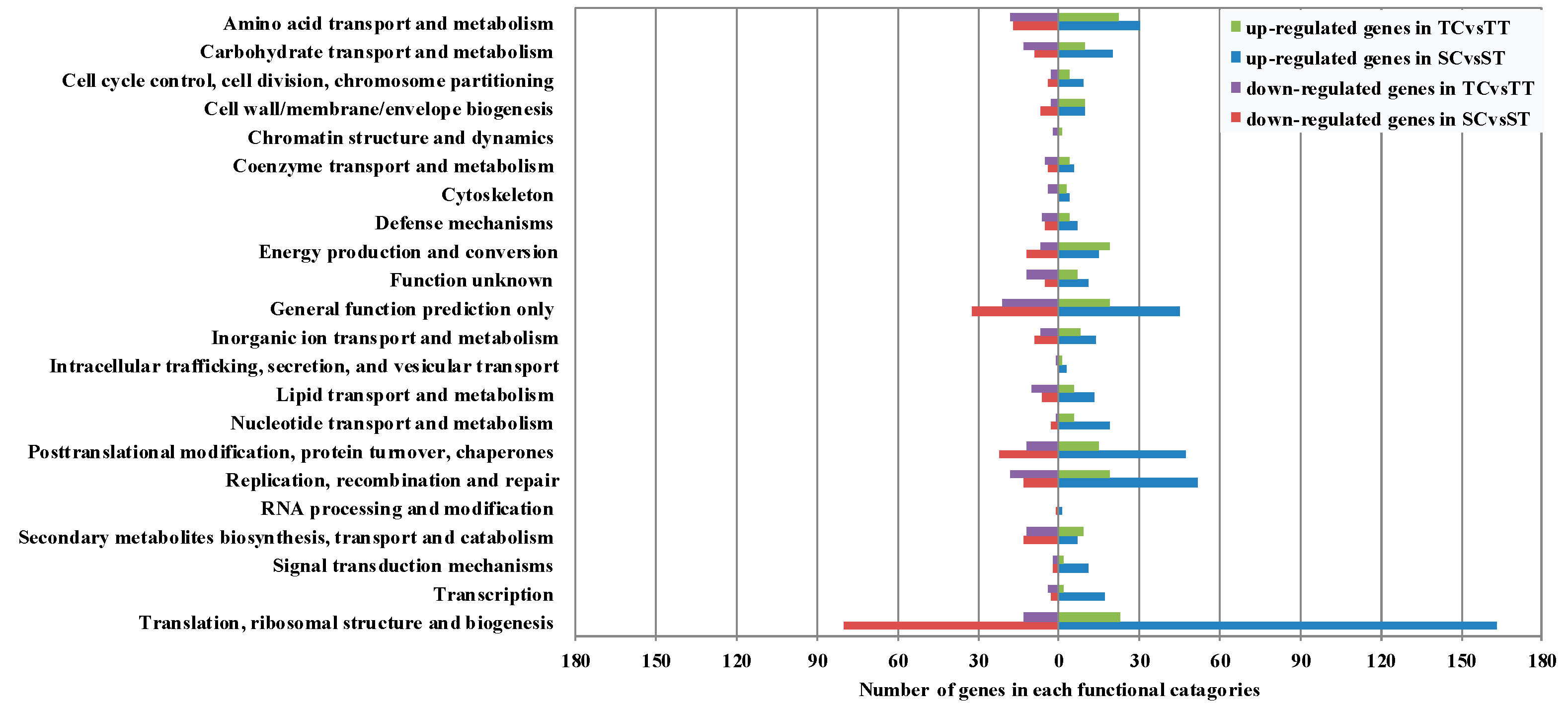
2.4. Digital Gene Expression Analysis


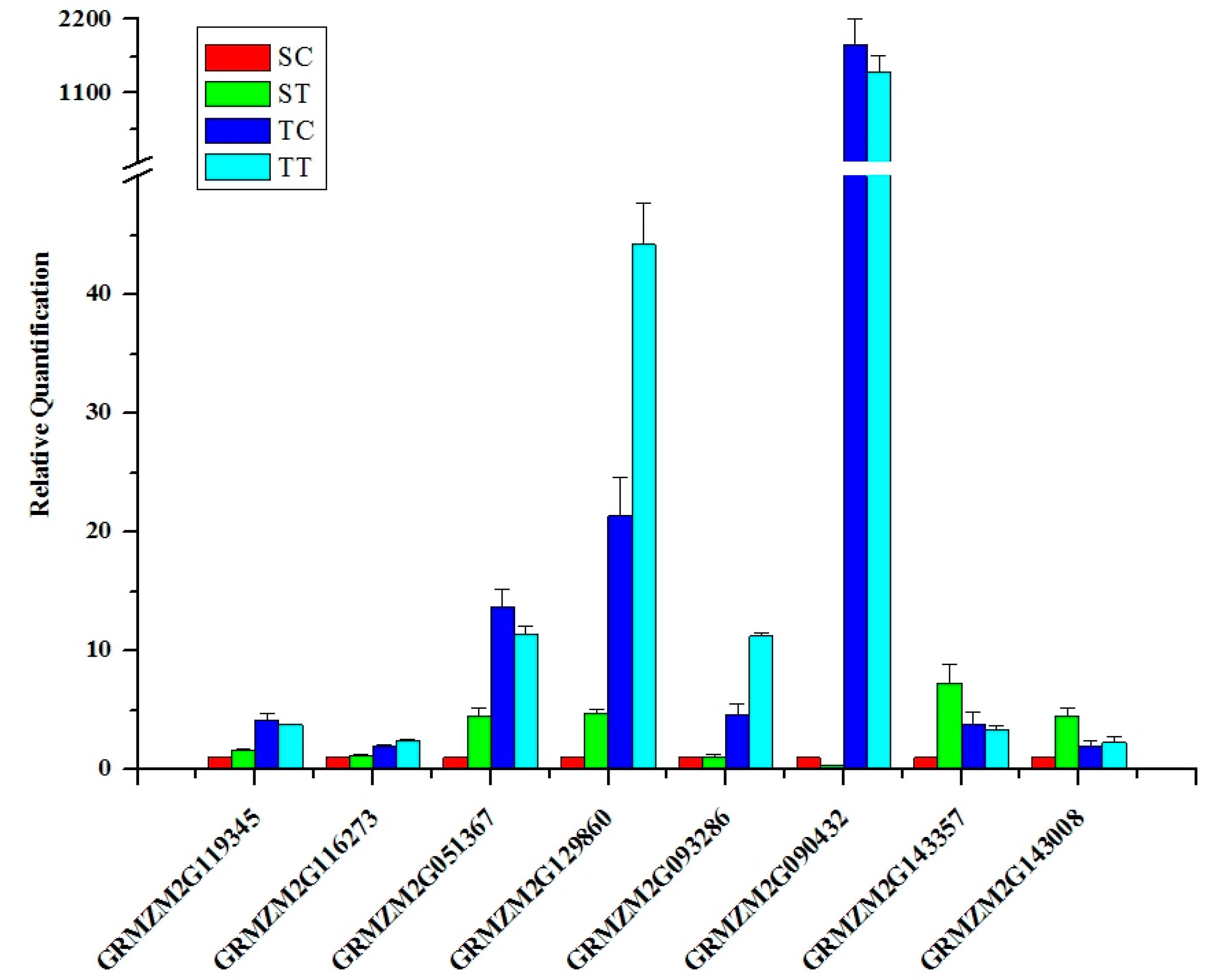
2.5. Searching and Validating of Potential Candidate Genes Involved in Nicosulfuron Metabolism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence ID | Annotation | fpkm | |||
|---|---|---|---|---|---|
| SC | ST | TC | TT | ||
| GRMZM2G119345 | ABC transporter protein | 12.6 | 28.3 | 27.0 | 40.7 |
| GRMZM2G116273 | glutathione S-transferase gene GST1 | 558.2 | 805.0 | 711.0 | 1019.3 |
| GRMZM2G330635 | glutathione S-transferase gene GSTU6 | 61.7 | 99.2 | 156.9 | 157.4 |
| GRMZM2G051367 | Glycosyltransferase | 8.9 | 18.1 | 22.6 | 24.8 |
| GRMZM2G370745 | cytochrome P450 monooxygenase, CYP72A28 | 18.8 | 28.0 | 26.2 | 64.7 |
| GRMZM2G022947 | cytochrome P450 monooxygenase, CYP727A4 | 12.7 | 25.3 | 19.2 | 25.9 |
| GRMZM2G129860 | cytochrome P450 monooxygenase, CYP72A5 | 3.0 | 7.7 | 24.6 | 25.2 |
| AC217947.4_FG002 | cytochrome P450 monooxygenase | 3.7 | 9.6 | 31.0 | 55.1 |
| GRMZM2G093286 | cytochrome P450 monooxygenase, CYP78A55 | 5.6 | 11.3 | 11.7 | 37.3 |
| GRMZM2G063756 | cytochrome P450 monooxygenase, CYP71C3v2 | 269.5 | 395.6 | 1131.4 | 1380.3 |
| GRMZM2G090432 | cytochrome P450 monooxygenase, CYP81A9 | 0 | 0.1 | 57.5 | 68.0 |
| GRMZM2G143357 | acetolactate synthase, AHAS109 | 4.2 | 24.5 | 5.5 | 6.3 |
| GRMZM2G143008 | acetolactate synthase, AHAS108 | 24.9 | 81.5 | 24.5 | 34.1 |

3. Experimental Section
3.1. Plant Material and RNA Extraction
3.2. cDNA Library Construction and RNA-Seq
3.3. RNA-Seq Reads Mapping and Transcript Assembly
3.4. Genes Annotation
3.5. Gene Validation and Expression Analysis
| Sequence ID | Forward Primer | Reverse Primer | Target Size bp |
|---|---|---|---|
| GRMZM2G119345 | AGGGTAGGATTCTGATGTTC | TGCTGATACTTCGGTCTGTTT | 73 |
| GRMZM2G116273 | GGGGAACCACCGACCAGAAAG | GCGTAGGGCGTAGCAAACAGG | 172 |
| GRMZM2G051367 | CGTTGCCTCCATCGCTTACTG | TGCCTGGTTCATTGGTCTCCC | 276 |
| GRMZM2G129860 | CGCCATCCTACACCCACG | TATGCGGTCAGTAACGAAA | 138 |
| GRMZM2G093286 | GGTTCGTGTTCGGCAAGGAG | GGGAAGTAGTCGCACAGGTT | 136 |
| GRMZM2G090432 | ACCACCCAACAGCCAAACCA | CCCAGGAGGTAGTGGAGCAA | 102 |
| GRMZM2G143357 | TGCTAAAGGGTTCAACATTCC | ACAGTCCTGCCATCACCATCC | 195 |
| GRMZM2G143008 | TTCTTCCTCGCCTCCTCTGGTC | ACAAAGCGTCGCAACTCCTCAC | 248 |
| β-actin | CATGGAGAACTGGCATCACACCTT | CTGCGTCATTTTCTCTCTGTTGGC | 118 |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shiferaw, B.; Prasanna, B.M.; Hellin, J.; Bänziger, M. Crops that feed the world 6. Past successes and future challenges to the role played by maize in global food security. Food Secur. 2011, 3, 307–327. [Google Scholar] [CrossRef]
- Zhang, Z.P. Development of chemical weed control and integrated weed management in China. Weed Biol. Manag. 2003, 3, 197–203. [Google Scholar] [CrossRef]
- Morton, C.A.; Harvey, R.G. Sweet corn (zea mays) hybrid tolerance to nicosulfuron. Weed Technol. 1992, 6, 91–96. [Google Scholar]
- Meyer, M.D.; Pataky, J.K.; Williams, M.M., II. Genetic factors influencing adverse effects of mesotrione and nicosulfuron on sweet corn yield. Agron. J. 2010, 102, 1139. [Google Scholar] [CrossRef]
- Fuentes, C.L.; Leroux, G.D. Rimsulfuron uptake, translocation, metabolism and als sensitivity to rimsulfuron in two maize hybrids. Agron. Colomb. 2003, 21, 17–27. [Google Scholar]
- Edwards, R.; Dixon, D.P.; Cummins, I.; Brazier-Hicks, M.; Skipsey, M. New perspectives on the metabolism and detoxification of synthetic compounds in plants. In Organic Xenobiotics and Plants; Springer: Berlin, Germany, 2011; pp. 125–148. [Google Scholar]
- Yuan, J.S.; Tranel, P.J.; Stewart, C.N. Non-target-site herbicide resistance: A family business. Trends Plant Sci. 2007, 12, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Saika, H.; Horita, J.; Taguchi-Shiobara, F.; Nonaka, S.; Ayako, N.-Y.; Iwakami, S.; Hori, K.; Matsumoto, T.; Tanaka, T.; Itoh, T. A novel rice cytochrome P450 gene, CYP72A31, confers tolerance to acetolactate synthase-inhibiting herbicides in rice and arabidopsis. Plant Physiol. 2014, 166, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Gion, K.; Inui, H.; Takakuma, K.; Yamada, T.; Kambara, Y.; Nakai, S.; Fujiwara, H.; Miyamura, T.; Imaishi, H.; Ohkawa, H. Molecular mechanisms of herbicide-inducible gene expression of tobacco CYP71AH11 metabolizing the herbicide chlorotoluron. Pestic. Biochem. Physiol. 2014, 108, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Siminszky, B. Plant cytochrome p450-mediated herbicide metabolism. Phytochem. Rev. 2006, 5, 445–458. [Google Scholar] [CrossRef]
- Pataky, J.K.; Williams, M.M., II; Riechers, D.E.; Meyer, M.D. A common genetic basis for cross-sensitivity to mesotrione and nicosulfuron in sweet corn hybrid cultivars and inbreds grown throughout north America. J. Am. Soc. Hortic. Sci. 2009, 134, 252–260. [Google Scholar]
- Pataky, J.K.; Meyer, M.D.; Bollman, J.D.; Boerboom, C.M.; Williams, M.M., II. Genetic basis for varied levels of injury to sweet corn hybrids from three cytochrome p450-metabolized herbicides. J. Am. Soc. Hortic. Sci. 2008, 133, 438–447. [Google Scholar]
- Barrett, M. Metabolism of herbicides by cytochrome P450 in corn. Drug Metabol. Drug Interact. 1995, 12, 299–316. [Google Scholar] [CrossRef] [PubMed]
- McGonigle, B.; Keeler, S.J.; Lau, S.-M.C.; Koeppe, M.K.; O’Keefe, D.P. A genomics approach to the comprehensive analysis of the glutathione S-transferase gene family in soybean and maize. Plant Physiol. 2000, 124, 1105–1120. [Google Scholar] [CrossRef] [PubMed]
- Sytykiewicz, H. Expression patterns of glutathione transferase gene (gsti) in maize seedlings under juglone-induced oxidative stress. Int. J. Mol. Sci. 2011, 12, 7982–7995. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.P.; Cummins, I.; Cole, D.J.; Edwards, R. Glutathione-mediated detoxification systems in plants. Curr. Opin. Plant Biol. 1998, 1, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Taylor, V.L.; Cummins, I.; Brazier-Hicks, M.; Edwards, R. Protective responses induced by herbicide safeners in wheat. Environ. Exp. Bot. 2013, 88, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Ghanashyam, C.; Bhattacharjee, A. Comprehensive expression analysis suggests overlapping and specific roles of rice glutathione S-transferase genes during development and stress responses. BMC Genomics 2010, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.; Duan, L.; Liu, Z.; Song, X.; Li, X.; Wang, C. Metolachlor-induced zmgt1 expression in maize cultivars is correlated with their tolerance to the herbicide. J. Food Agric. Environ. 2012, 10, 621–623. [Google Scholar]
- Pang, S.; Duan, L.; Liu, Z.; Song, X.; Li, X.; Wang, C. Co-induction of a glutathione-S-transferase, a glutathione transporter and an abc transporter in maize by xenobiotics. PLoS One 2012, 7, e40712. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.; Ran, Z.; Liu, Z.; Song, X.; Duan, L.; Li, X.; Wang, C. Enantioselective induction of a glutathione-S-transferase, a glutathione transporter and an abc transporter in maize by metolachlor and its (S)-isomer. PLoS One 2012, 7, e48085. [Google Scholar] [CrossRef] [PubMed]
- Schuman, B.; Alfaro, J.A.; Evans, S.V. Glycosyltransferase structure and function. In Bioactive Conformation I; Springer: Berlin, Germany, 2007; pp. 217–257. [Google Scholar]
- Kreuz, K.; Tommasini, R.; Martinoia, E. Old enzymes for a new job (herbicide detoxification in plants). Plant Physiol. 1996, 111, 349. [Google Scholar] [PubMed]
- Brazier, M.; Cole, D.J.; Edwards, R. O-glucosyltransferase activities toward phenolic natural products and xenobiotics in wheat and herbicide-resistant and herbicide-susceptible black-grass (alopecurus myosuroides). Phytochemistry 2002, 59, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Forlani, G.; Nielsen, E.; Landi, P.; Tuberosa, R. Chlorsulfuron tolerance and acetolactate synthase activity in corn (Zea mays L.) inbred lines. Weed Sci. 1991, 39, 553–557. [Google Scholar]
- Carey, J.B.; Penner, D.; Kells, J.J. Physiological basis for nicosulfuron and primisulfuron selectivity in five plant species. Weed Sci. 1997, 45, 22–30. [Google Scholar]
- Pataky, J.K.; Nordby, J.N.; Williams, M.M.; Riechers, D.E. Inheritance of cross-sensitivity in sweet corn to herbicides applied postemergence. J. Am. Soc. Hortic. Sci. 2006, 131, 744–751. [Google Scholar]
- Zhu, T.; Mettenburg, K.; Peterson, D.J.; Tagliani, L.; Baszczynski, C.L. Engineering herbicide-resistant maize using chimeric RNA/DNA oligonucleotides. Nat. Biotechnol. 2000, 18, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Tao, B.; Qiu, L.; Jin, L.; Wu, J. Role of physiological mechanisms and EPSPS gene expression in glyphosate resistance in wild soybeans (Glycine soja). Pestic. Biochem. Physiol. 2014, 109, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Sala, C.A.; Bulos, M. Inheritance and molecular characterization of broad range tolerance to herbicides targeting acetohydroxyacid synthase in sunflower. Theor. Appl. Genet. 2012, 124, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Thyssen, G.; McCarty, J.C.; Li, P.; Jenkins, J.N.; Fang, D.D. Genetic mapping of non-target-site resistance to a sulfonylurea herbicide (envoke®) in upland cotton (Gossypium hirsutum L.). Mol. Breed. 2014, 33, 341–348. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Riggins, C.W.; Peng, Y.; Stewart, C.N.; Tranel, P.J. Characterization of de novo transcriptome for waterhemp (Amaranthus tuberculatus) using GS-FLX 454 pyrosequencing and its application for studies of herbicide target-site genes. Pest Manag. Sci. 2010, 66, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, X.-Y.; Li, Y.-F. De novo assembly and characterization of the barnyardgrass (Echinochloa crus-galli) transcriptome using next-generation pyrosequencing. PLoS One 2013, 8, e69168. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Shen, X.; Ma, Q.; Yang, C.; Liu, S.; Chen, Y. Transcriptome profiling to discover putative genes associated with paraquat resistance in goosegrass (Eleusine indica L.). PLoS One 2014, 9, e99940. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2010, 26, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The kegg resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. Ebseq: An empirical bayes hierarchical model for inference in RNA-Seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Delye, C. Unravelling the genetic bases of non-target-site-based resistance (NTSR) to herbicides: A major challenge for weed science in the forthcoming decade. Pest Manag. Sci. 2013, 69, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yu, H.; Zhang, F.; Lin, C.; Gao, J.; Fang, J.; Ding, X.; Shen, Z.; Xu, X. A built-in strategy to mitigate transgene spreading from genetically modified corn. PLoS One 2013, 8, e81645. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Wang, X.; Ren, T. Expression of a wheat cytochrome P450 monooxygenase cDNA in yeast catalyzes the metabolism of sulfonylurea herbicides. Pestic. Biochem. Physiol. 2006, 85, 1–6. [Google Scholar] [CrossRef]
- Pan, G.; Zhang, X.; Liu, K.; Zhang, J.; Wu, X.; Zhu, J.; Tu, J. Map-based cloning of a novel rice cytochrome p450 gene CYP81A6 that confers resistance to two different classes of herbicides. Plant Mol. Biol. 2006, 61, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Karavangeli, M.; Labrou, N.E.; Clonis, Y.D.; Tsaftaris, A. Development of transgenic tobacco plants overexpressing maize glutathione S-transferase I for chloroacetanilide herbicides phytoremediation. Biomol. Eng. 2005, 22, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Yang, S.N.; Zhang, J.J.; Zhang, J.J.; Tan, L.R.; Yang, H. A collection of glycosyltransferases from rice (Oryza sativa) exposed to atrazine. Gene 2013, 531, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Keeler, S.J.; Sanders, P.; Smith, J.K.; Mazur, B.J. Regulation of tobacco acetolactate synthase gene expression. Plant Physiol. 1993, 102, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Ghio, C.; Ramos, M.L.; Altieri, E.; Bulos, M.; Sala, C.A. Molecular characterization of Als1, an acetohydroxyacid synthase mutation conferring resistance to sulfonylurea herbicides in soybean. Theor. Appl. Genet. 2013, 126, 2957–2968. [Google Scholar] [CrossRef] [PubMed]
- Iwakami, S.; Uchino, A.; Watanabe, H.; Yamasue, Y.; Inamura, T. Isolation and expression of genes for acetolactate synthase and acetyl-CoA carboxylase in Echinochloa phyllopogon, a polyploid weed species. Pest Manag. Sci. 2012, 68, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Galli, V.; da Silva Messias, R.; dos Anjos e Silva, S.D.; Rombaldi, C.V. Selection of reliable reference genes for quantitative real-time polymerase chain reaction studies in maize grains. Plant Cell Rep. 2013, 32, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. Tophat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Xu, X.; Li, B.; Wang, X.; Wang, G.; Li, M. RNA-Seq Transcriptome Analysis of Maize Inbred Carrying Nicosulfuron-Tolerant and Nicosulfuron-Susceptible Alleles. Int. J. Mol. Sci. 2015, 16, 5975-5989. https://doi.org/10.3390/ijms16035975
Liu X, Xu X, Li B, Wang X, Wang G, Li M. RNA-Seq Transcriptome Analysis of Maize Inbred Carrying Nicosulfuron-Tolerant and Nicosulfuron-Susceptible Alleles. International Journal of Molecular Sciences. 2015; 16(3):5975-5989. https://doi.org/10.3390/ijms16035975
Chicago/Turabian StyleLiu, Xiaomin, Xian Xu, Binghua Li, Xueqing Wang, Guiqi Wang, and Moran Li. 2015. "RNA-Seq Transcriptome Analysis of Maize Inbred Carrying Nicosulfuron-Tolerant and Nicosulfuron-Susceptible Alleles" International Journal of Molecular Sciences 16, no. 3: 5975-5989. https://doi.org/10.3390/ijms16035975
APA StyleLiu, X., Xu, X., Li, B., Wang, X., Wang, G., & Li, M. (2015). RNA-Seq Transcriptome Analysis of Maize Inbred Carrying Nicosulfuron-Tolerant and Nicosulfuron-Susceptible Alleles. International Journal of Molecular Sciences, 16(3), 5975-5989. https://doi.org/10.3390/ijms16035975