
Conformational Analysis of Misfolded Protein Aggregation by FRET and Live-Cell Imaging Techniques



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Spectroscopic Imaging for Elucidation of the Mechanism of Proteostasis
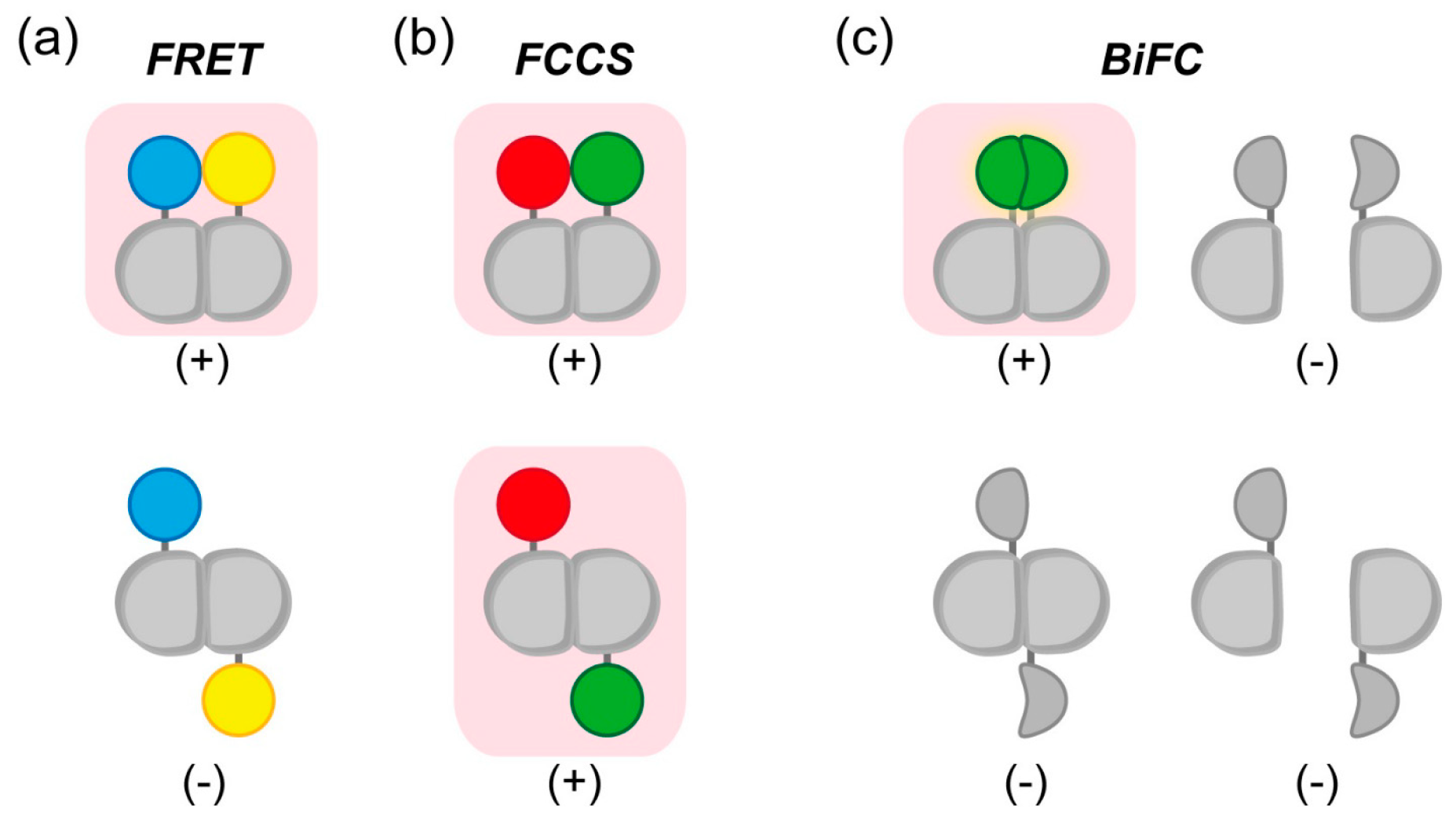
2.1. Detection of Interactions between Aggregation-Prone Proteins and Molecular Chaperones by Förster/Fluorescence Resonance Energy Transfer (FRET) and Fluorescence Correlation Spectroscopy (FCS)
2.1.1. Using FRET to Elucidate the Association between a Chaperone and polyQ Protein
2.1.2. Detection of Soluble Oligomers of Expanded polyQ and Mutant Superoxide Dismutase 1 (SOD1) by FCS
2.1.3. Efficient and Reliable Fluorescent Proteins for Use in FLIM-FRET Analysis
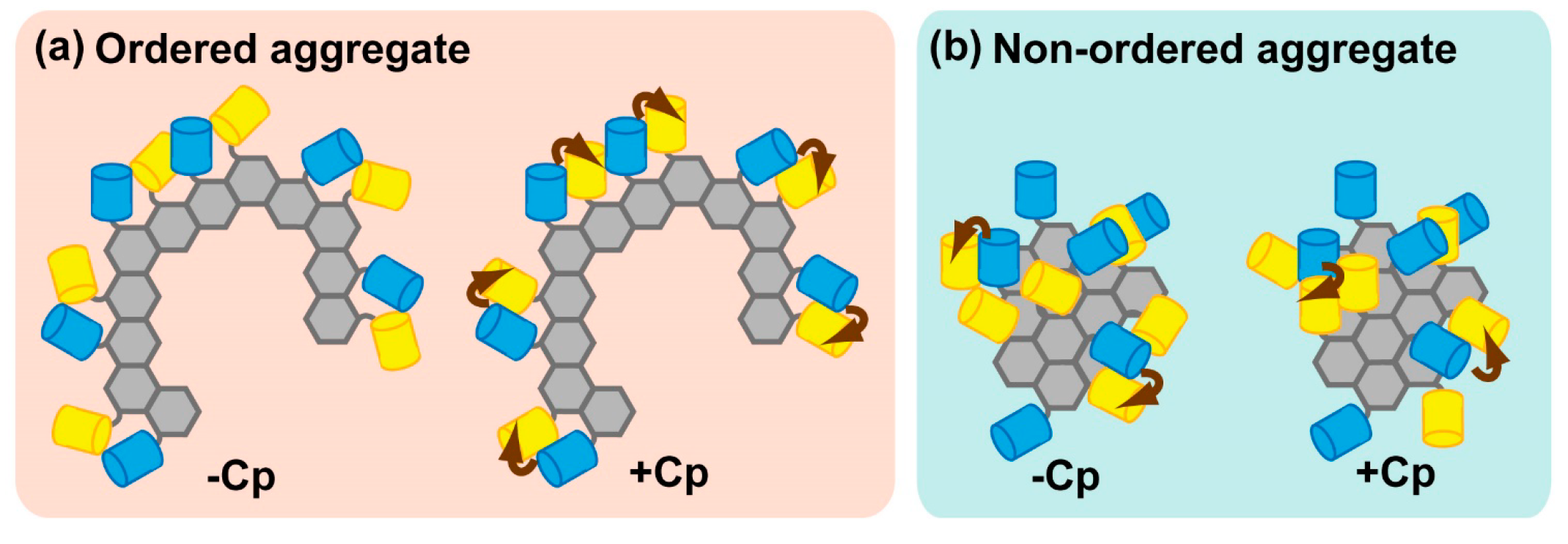
2.2. Characterization of Inclusion Bodies Containing Aggregation-Prone Proteins by FRET
2.2.1. FRET of Sequestered Aggregation-Prone Proteins in Inclusion Bodies

2.2.2. Importance of the Orientation Factor in FRET, and Introduction of Circularly Permutated Fluorescent Proteins

2.2.3. Physiological Relevance of Ordered Sequestration of Mutant SOD1 in Inclusion Bodies
2.3. Dimerization Detection of ALS-Associated SOD1 Protein
2.4. Biosensors to Monitor Physiological Reactions and States Related to Proteostasis in Cells and Tissues
2.4.1. Indicator for Real-Time Detection of Apoptosis by FRET or FCCS

2.4.2. Direct Measurements of Dissociation Constants in Living Cells
2.4.3. Calcium and Redox Sensors
2.4.4. Fluorescent or Luminescent Reporters for Misfolding and Stress Responses.
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [PubMed]
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008, 22, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Van Oosten-Hawle, P.; Morimoto, R.I. Organismal proteostasis: Role of cell-nonautonomous regulation and transcellular chaperone signaling. Genes Dev. 2014, 28, 1533–1543. [Google Scholar]
- Mardones, P.; Martinez, G.; Hetz, C. Control of systemic proteostasis by the nervous system. Trends Cell Biol. 2015, 25, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; del Tredici, K.; Lee, V.M.; Trojanowski, J.Q. Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat. Rev. Neurosci. 2015, 16, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Zempel, H.; Mandelkow, E. Lost after translation: Missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014, 37, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Ann. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O'Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- The Huntington’s disease collaborative research group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar]
- Trottier, Y.; Lutz, Y.; Stevanin, G.; Imbert, G.; Devys, D.; Cancel, G.; Saudou, F.; Weber, C.; David, G.; Tora, L.; et al. Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature 1995, 378, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Geetha, T.; Babu, J.R. Failure of ubiquitin proteasome system: Risk for neurodegenerative diseases. Neurodegener. Dis. 2014, 14, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Brundin, P. The ubiquitin proteasome system in neurodegenerative diseases: Sometimes the chicken, sometimes the egg. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef] [PubMed]
- Munishkina, L.A.; Fink, A.L. Fluorescence as a method to reveal structures and membrane-interactions of amyloidogenic proteins. Biochim. Biophys. Acta 2007, 1768, 1862–1885. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A.; Tsien, R.Y. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 2000, 327, 472–500. [Google Scholar] [PubMed]
- Rigler, R.; Mets, U.; Widengren, J.; Kask, P. Fluorescence correlation spectroscopy with high count rate and low-background—Analysis of translational diffusion. Eur. Biophys. J. Biophys. 1993, 22, 169–175. [Google Scholar] [CrossRef]
- Lippincott-Schwartz, J.; Snapp, E.; Kenworthy, A. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2001, 2, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Bacia, K.; Kim, S.A.; Schwille, P. Fluorescence cross-correlation spectroscopy in living cells. Nat. Methods 2006, 3, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Wanker, E.E. Protein aggregation and pathogenesis of Huntington's disease: Mechanisms and correlations. Biol. Chem. 2000, 381, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Muchowski, P.J.; Schaffar, G.; Sittler, A.; Wanker, E.E.; Hayer-Hartl, M.K.; Hartl, F.U. Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2000, 97, 7841–7846. [Google Scholar] [CrossRef] [PubMed]
- Schaffar, G.; Breuer, P.; Boteva, R.; Behrends, C.; Tzvetkov, N.; Strippel, N.; Sakahira, H.; Siegers, K.; Hayer-Hartl, M.; Hartl, F.U. Cellular toxicity of polyglutamine expansion proteins: Mechanism of transcription factor deactivation. Mol. Cell 2004, 15, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Kikuchi, S.; Katada, S.; Nagai, Y.; Nishizawa, M.; Onodera, O. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum. Mol. Genet. 2008, 17, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Behrends, C.; Langer, C.A.; Boteva, R.; Bottcher, U.M.; Stemp, M.J.; Schaffar, G.; Rao, B.V.; Giese, A.; Kretzschmar, H.; Siegers, K.; et al. Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol. Cell 2006, 23, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Kubota, H.; Pack, C.G.; Matsumoto, G.; Hirayama, S.; Takahashi, Y.; Kimura, H.; Kinjo, M.; Morimoto, R.I.; Nagata, K. Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat. Cell Biol. 2006, 8, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.; Geller, R.; Spiess, C.; Frydman, J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat. Cell Biol. 2006, 8, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, E.; Zako, T.; Muto, H.; Itoo, Y.; Sorgjerd, K.; Terada, N.; Abe, A.; Miyazawa, M.; Kitamura, A.; Kitaura, H.; et al. Prefoldin protects neuronal cells from polyglutamine toxicity by preventing aggregation formation. J. Biol. Chem. 2013, 288, 19958–19972. [Google Scholar] [CrossRef] [PubMed]
- Rothlein, C.; Miettinen, M.S.; Borwankar, T.; Burger, J.; Mielke, T.; Kumke, M.U.; Ignatova, Z. Architecture of polyglutamine-containing fibrils from time-resolved fluorescence decay. J. Biol. Chem. 2014, 289, 26817–26828. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Okamoto, Y.; Popiel, H.A.; Fujikake, N.; Toda, T.; Kinjo, M.; Nagai, Y. Detection of polyglutamine protein oligomers in cells by fluorescence correlation spectroscopy. J. Biol. Chem. 2007, 282, 24039–24048. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Inada, N.; Kubota, H.; Matsumoto, G.; Kinjo, M.; Morimoto, R.I.; Nagata, K. Dysregulation of the proteasome increases the toxicity of ALS-linked mutant SOD1. Genes Cells 2014, 19, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Rohde, G.; Eckermann, K.; Sroka, K.; Schaefer, M.K.; Dohm, C.P.; Kermer, P.; Haase, G.; Wouters, F.; Bahr, M.; et al. Mutant SOD1 detoxification mechanisms in intact single cells. Cell Death Differ. 2008, 15, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, R. Imaging spatiotemporal dynamics of neuronal signaling using fluorescence resonance energy transfer and fluorescence lifetime imaging microscopy. Curr. Opin. Neurobiol. 2006, 16, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Zhai, S.; Ark, E.D.; Parra-Bueno, P.; Yasuda, R. Long-distance integration of nuclear ERK signaling triggered by activation of a few dendritic spines. Science 2013, 342, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Padilla-Parra, S.; Auduge, N.; Lalucque, H.; Mevel, J.C.; Coppey-Moisan, M.; Tramier, M. Quantitative comparison of different fluorescent protein couples for fast FRET-FLIM acquisition. Biophys. J. 2009, 97, 2368–2376. [Google Scholar] [CrossRef] [PubMed]
- Tramier, M.; Zahid, M.; Mevel, J.C.; Masse, M.J.; Coppey-Moisan, M. Sensitivity of CFP/YFP and GFP/mCherry pairs to donor photobleaching on FRET determination by fluorescence lifetime imaging microscopy in living cells. Microsc. Res. Tech. 2006, 69, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Murakoshi, H.; Lee, S.J.; Yasuda, R. Highly sensitive and quantitative FRET-FLIM imaging in single dendritic spines using improved non-radiative YFP. Brain Cell Biol. 2008, 36, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Goedhart, J.; von Stetten, D.; Noirclerc-Savoye, M.; Lelimousin, M.; Joosen, L.; Hink, M.A.; van Weeren, L.; Gadella, T.W., Jr.; Royant, A. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 2012, 3, 751. [Google Scholar] [CrossRef] [PubMed]
- Erard, M.; Fredj, A.; Pasquier, H.; Beltolngar, D.B.; Bousmah, Y.; Derrien, V.; Vincent, P.; Merola, F. Minimum set of mutations needed to optimize cyan fluorescent proteins for live cell imaging. Mol. Biosyst. 2013, 9, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.L.; Kim, H.; Markwardt, M.L.; Chen, L.; Fromme, R.; Rizzo, M.A.; Wachter, R.M. The 1.6 Å resolution structure of a FRET-optimized cerulean fluorescent protein. Acta Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 767–773. [Google Scholar] [CrossRef]
- Baker, M.J.; Tatsuta, T.; Langer, T. Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Bartoszewska, M.; Williams, C.; Kikhney, A.; Opalinski, L.; van Roermund, C.W.; de Boer, R.; Veenhuis, M.; van der Klei, I.J. Peroxisomal proteostasis involves a Lon family protein that functions as protease and chaperone. J. Biol. Chem. 2012, 287, 27380–27395. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R.; Ishimoto, Y.; Nangaku, M. Proteostasis in endoplasmic reticulum--new mechanisms in kidney disease. Nat. Rev. Nephrol. 2014, 10, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Avezov, E.; Konno, T.; Zyryanova, A.; Chen, W.; Laine, R.; Crespillo-Casado, A.; Melo, E.; Ushioda, R.; Nagata, K.; Kaminski, C.F.; et al. Retarded PDI diffusion and a reductive shift in poise of the calcium depleted endoplasmic reticulum. BMC Biol. 2015, 13. [Google Scholar] [CrossRef]
- Kitamura, A.; Kubota, H. Amyloid oligomers: Dynamics and toxicity in the cytosol and nucleus. FEBS J. 2010, 277, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008, 454, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Kedersha, N. RNA granules. J. Cell Biol. 2006, 172, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Escusa-Toret, S.; Vonk, W.I.; Frydman, J. Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat. Cell Biol. 2013, 15, 1231–1243. [Google Scholar] [CrossRef] [PubMed]
- Stefl, R.; Skrisovska, L.; Allain, F.H. RNA sequence- and shape-dependent recognition by proteins in the ribonucleoprotein particle. EMBO Rep. 2005, 6, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Latonen, L. Nucleolar aggresomes as counterparts of cytoplasmic aggresomes in proteotoxic stress: Proteasome inhibitors induce nuclear ribonucleoprotein inclusions that accumulate several key factors of neurodegenerative diseases and cancer. BioEssays 2011, 33, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Morimoto, R.I. How the nucleus copes with proteotoxic stress. Cur. Biol. 2014, 24, R463–R474. [Google Scholar] [CrossRef]
- Kim, S.; Nollen, E.A.; Kitagawa, K.; Bindokas, V.P.; Morimoto, R.I. Polyglutamine protein aggregates are dynamic. Nat. Cell Biol. 2002, 4, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, G.; Kim, S.; Morimoto, R.I. Huntingtin and mutant SOD1 form aggregate structures with distinct molecular properties in human cells. J. Biol. Chem. 2006, 281, 4477–4485. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.J.; Lyakhovetsky, R.; Werdiger, A.C.; Gitler, A.D.; Soen, Y.; Kaganovich, D. Compartmentalization of superoxide dismutase 1 (SOD1G93A) aggregates determines their toxicity. Proc. Natl. Acad. Sci. USA 2012, 109, 15811–15816. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A. Visualization of the spatial and temporal dynamics of intracellular signaling. Dev. Cell 2003, 4, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Yamada, S.; Tominaga, T.; Ichikawa, M.; Miyawaki, A. Expanded dynamic range of fluorescent indicators for Ca2+ by circularly permuted yellow fluorescent proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 10554–10559. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Biancalana, M.; Koide, S.; Shea, J.E. Binding modes of Thioflavin-T to the single-layer beta-sheet of the peptide self-assembly mimics. J. Mol. Biol. 2009, 394, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Hao, R.; Nanduri, P.; Rao, Y.; Panichelli, R.S.; Ito, A.; Yoshida, M.; Yao, T.P. Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol. Cell 2013, 51, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Simoes-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.A.; Cuendet, M. HDAC6 as a target for neurodegenerative diseases: What makes it different from the other HDACs? Mol. Neurodegener. 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Torres, A.S.; O'Halloran, T.V. Oxygen-induced maturation of SOD1: A key role for disulfide formation by the copper chaperone ccs. EMBO J. 2004, 23, 2872–2881. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; O’Halloran, T.V. Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo- and reduced form of SOD1, leading to unfolding and oxidative aggregation. J. Biol. Chem. 2005, 280, 17266–17274. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.S.; Antonyuk, S.V.; Kershaw, N.M.; Strange, R.W.; Samar Hasnain, S. Ligand binding and aggregation of pathogenic SOD1. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Ray, S.S.; Nowak, R.J.; Strokovich, K.; Brown, R.H., Jr.; Walz, T.; Lansbury, P.T., Jr. An intersubunit disulfide bond prevents in vitro aggregation of a superoxide dismutase-1 mutant linked to familial amytrophic lateral sclerosis. Biochemistry 2004, 43, 4899–4905. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, H.; Lee, J.H.; Kwon, D.Y.; Genovesio, A.; Fenistein, D.; Ogier, A.; Brondani, V.; Grailhe, R. Dimerization, oligomerization, and aggregation of human amyotrophic lateral sclerosis Copper/Zinc superoxide dismutase 1 protein mutant forms in live cells. J. Biol. Chem. 2014, 289, 15094–15103. [Google Scholar] [CrossRef] [PubMed]
- Kerppola, T.K. Complementary methods for studies of protein interactions in living cells. Nat. Methods 2006, 3, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Tyas, L.; Brophy, V.A.; Pope, A.; Rivett, A.J.; Tavare, J.M. Rapid caspase-3 activation during apoptosis revealed using fluorescence-resonance energy transfer. EMBO Rep. 2000, 1, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.Q.; Yu, V.C.; Pu, Y.; Chang, D.C. Application of the fluorescence resonance energy transfer method for studying the dynamics of caspase-3 activation during UV-induced apoptosis in living HeLa cells. Biochem. Biophys. Res. Commun. 2001, 283, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Rehm, M.; Dussmann, H.; Janicke, R.U.; Tavare, J.M.; Kogel, D.; Prehn, J.H. Single-cell fluorescence resonance energy transfer analysis demonstrates that caspase activation during apoptosis is a rapid process. Role of caspase-3. J. Biol. Chem. 2002, 277, 24506–24514. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, K.; Nagai, T.; Miyawaki, A.; Miura, M. Spatio-temporal activation of caspase revealed by indicator that is insensitive to environmental effects. J. Cell Biol. 2003, 160, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Miyawaki, A. A high-throughput method for development of FRET-based indicators for proteolysis. Biochem. Biophys. Res. Commun. 2004, 319, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Shcherbakova, D.M.; Hink, M.A.; Joosen, L.; Gadella, T.W.; Verkhusha, V.V. An orange fluorescent protein with a large stokes shift for single-excitation multicolor FCCS and FRET imaging. J. Am. Chem. Soc. 2012, 134, 7913–7923. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A.; Shcherbakova, D.M.; Verkhusha, V.V. Red fluorescent proteins: Chromophore formation and cellular applications. Curr. Opin. Struct. Biol. 2012, 22, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Wada, I.; Tamura, M.; Kinjo, M. Direct detection of caspase-3 activation in single live cells by cross-correlation analysis. Biochem. Biophys. Res. Commun. 2004, 324, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Halfmann, R.; Alberti, S.; Lindquist, S. Prions, protein homeostasis, and phenotypic diversity. Trends Cell Biol. 2010, 20, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyama, B.H.; Weissman, J.S. Amyloid structure: Conformational diversity and consequences. Ann. Rev. Biochem. 2011, 80, 557–585. [Google Scholar] [CrossRef] [PubMed]
- Ohta, S.; Kawai-Noma, S.; Kitamura, A.; Pack, C.G.; Kinjo, M.; Taguchi, H. The interaction of Hsp104 with yeast prion Sup35 as analyzed by fluorescence cross-correlation spectroscopy. Biochem. Biophys. Res. Commun. 2013, 442, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Vukojevic, V.; Papadopoulos, D.K.; Terenius, L.; Gehring, W.J.; Rigler, R. Quantitative study of synthetic Hox transcription factor-DNA interactions in live cells. Proc. Natl. Acad. Sci. USA 2010, 107, 4093–4098. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, M.; Mikuni, S.; Muto, H.; Kinjo, M. Determination of dissociation constant of the nfkappab p50/p65 heterodimer using fluorescence cross-correlation spectroscopy in the living cell. Biochem. Biophys. Res. Commun. 2013, 436, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Sadaie, W.; Harada, Y.; Matsuda, M.; Aoki, K. Quantitative in vivo fluorescence cross-correlation analyses highlight the importance of competitive effects in the regulation of protein-protein interactions. Mol. Cell. Biol. 2014, 34, 3272–3290. [Google Scholar] [CrossRef] [PubMed]
- Ong, D.S.; Mu, T.W.; Palmer, A.E.; Kelly, J.W. Endoplasmic reticulum Ca2+ increases enhance mutant glucocerebrosidase proteostasis. Nat. Chem. Biol. 2010, 6, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Nagata, K. Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 2011, 3, a007526. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, M. Genetically encoded probes for measurement of intracellular calcium. Methods Cell Biol. 2010, 99, 153–182. [Google Scholar] [PubMed]
- Meyer, A.J.; Dick, T.P. Fluorescent protein-based redox probes. Antioxid. Redox Signal. 2010, 13, 621–650. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Oku, M.; Akeyama, N.; Itoyama, A.; Yurimoto, H.; Kuge, S.; Fujiki, Y.; Sakai, Y. A novel fluorescent sensor protein for visualization of redox states in the cytoplasm and in peroxisomes. Mol. Cell. Biol. 2010, 30, 3758–3766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomosugi, W.; Matsuda, T.; Tani, T.; Nemoto, T.; Kotera, I.; Saito, K.; Horikawa, K.; Nagai, T. An ultramarine fluorescent protein with increased photostability and pH insensitivity. Nat. Methods 2009, 6, 351–353. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, K.; Nagai, T.; Nakano, M.; Ichinose, H.; Nakabayashi, T.; Ohta, N.; Hisabori, T. Redox sensor proteins for highly sensitive direct imaging of intracellular redox state. Biochem. Biophys. Res. Commun. 2015, 457, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, S.; Avezov, E.; Zyryanova, A.; Konno, T.; Mendes-Silva, L.; Melo, E.P.; Harding, H.P.; Ron, D. Intact protein folding in the glutathione-depleted endooplasmic reticulum implicates alternative protein thiol reductant. eLife 2014, 3, e03421. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Kasturi, P.; Bracher, A.; Loew, C.; Zheng, M.; Villella, A.; Garza, D.; Hartl, F.U.; Raychaudhuri, S. Firefly luciferase mutants as sensors of proteome stress. Nat. Methods 2011, 8, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Geiler-Samerotte, K.A.; Dion, M.F.; Budnik, B.A.; Wang, S.M.; Hartl, D.L.; Drummond, D.A. Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc. Natl. Acad. Sci. USA 2011, 108, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, G.; Stojanovic, A.; Holmberg, C.I.; Kim, S.; Morimoto, R.I. Structural properties and neuronal toxicity of amyotrophic lateral sclerosis-associated Cu/Zn superoxide dismutase 1 aggregates. J. Cell Biol. 2005, 171, 75–85. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitamura, A.; Nagata, K.; Kinjo, M. Conformational Analysis of Misfolded Protein Aggregation by FRET and Live-Cell Imaging Techniques. Int. J. Mol. Sci. 2015, 16, 6076-6092. https://doi.org/10.3390/ijms16036076
Kitamura A, Nagata K, Kinjo M. Conformational Analysis of Misfolded Protein Aggregation by FRET and Live-Cell Imaging Techniques. International Journal of Molecular Sciences. 2015; 16(3):6076-6092. https://doi.org/10.3390/ijms16036076
Chicago/Turabian StyleKitamura, Akira, Kazuhiro Nagata, and Masataka Kinjo. 2015. "Conformational Analysis of Misfolded Protein Aggregation by FRET and Live-Cell Imaging Techniques" International Journal of Molecular Sciences 16, no. 3: 6076-6092. https://doi.org/10.3390/ijms16036076
APA StyleKitamura, A., Nagata, K., & Kinjo, M. (2015). Conformational Analysis of Misfolded Protein Aggregation by FRET and Live-Cell Imaging Techniques. International Journal of Molecular Sciences, 16(3), 6076-6092. https://doi.org/10.3390/ijms16036076