
Understanding FRET as a Research Tool for Cellular Studies



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background
2. Introduction


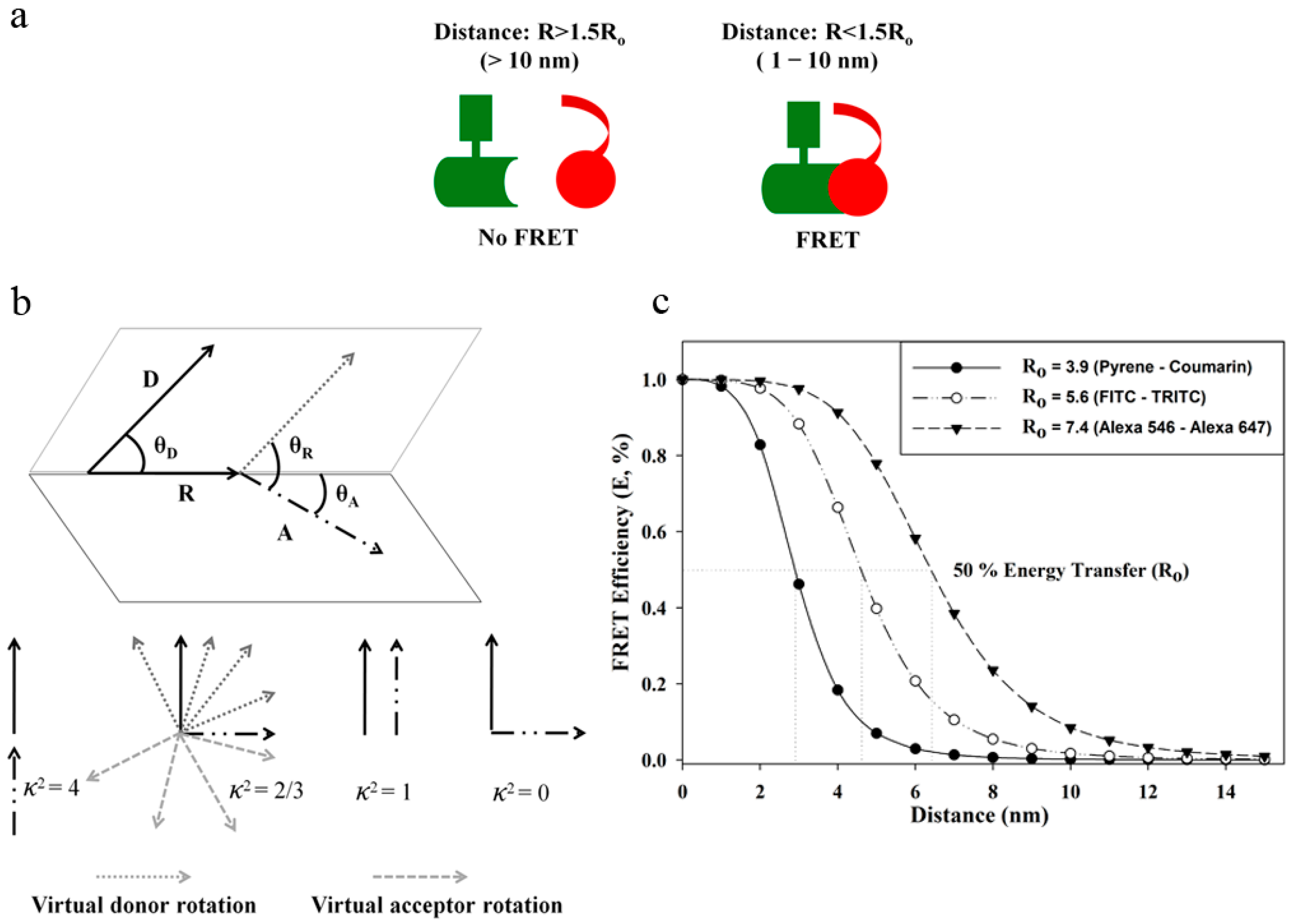
3. FRET: An Index for Sub-10 Nanometer Distances
4. Lighting up Molecules for FRET
4.1. An Approach Based on Fluorescent Affinity Reagents
4.2. An Approach Based on Fluorescent Proteins
4.3. An Approach Based on Bioorthogonal Chemistry
5. Is Your Instrument FRET Friendly?
6. Methods for Measuring FRET


6.1. Fluorescence Intensity Based Approach
6.1.1. Donor Quenching Method
6.1.2. Acceptor Photobleaching Method
6.1.3. Sensitized Acceptor Excitation Method
Two-Channel Emission or Excitation Ratio Measurement
Three-Channel Emission Measurement
Spectral Analysis for FRET
6.1.4. Donor Photobleaching Method
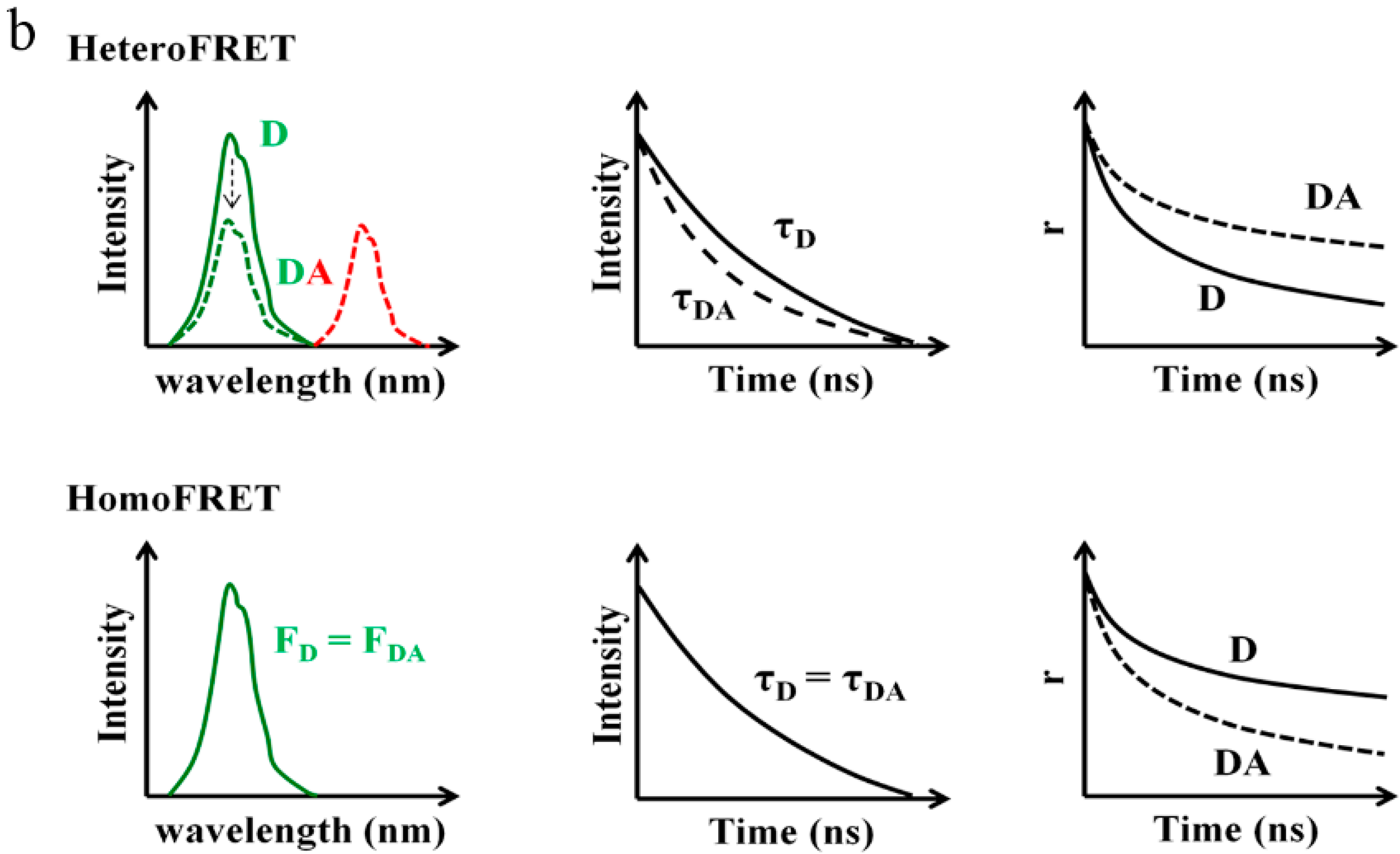
6.2. Fluorescence Lifetime Based Approach
6.3. Fluorescence Anisotropy Based Approach
7. Applications of FRET in Membrane Biology
7.1. Organization of Antigen Presenting Molecules in the Plasma Membrane of B Cells
7.2. Cytokine Receptors and MHC Proteins in T Cells
7.3. Dynamic Reorganization of Membrane Proteins in T Cells during Immune Synapse Formation
7.4. Elucidating the Membrane Features of ErbB/HER Kinases
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cooper, G.M.; Hausman, R.E. The molecular composition of cells. In The Cell: A Molecular Approach, 4th ed.; ASM Press: Washington, DC, USA, 2007; pp. 43–72. [Google Scholar]
- Cebecauer, M.; Spitaler, M.; Serge, A.; Magee, A.I. Signalling complexes and clusters: Functional advantages and methodological hurdles. J. Cell Sci. 2010, 123, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Miermont, A.; Waharte, F.; Hu, S.; McClean, M.N.; Bottani, S.; Leon, S.; Hersen, P. Severe osmotic compression triggers a slowdown of intracellular signaling, which can be explained by molecular crowding. Proc. Natl. Acad. Sci. USA 2013, 110, 5725–5730. [Google Scholar] [CrossRef] [PubMed]
- Masters, B. Paths to Förster resonance energy transfer (FRET) theory. Eur. Phys. J. H 2014, 39, 87–139. [Google Scholar] [CrossRef]
- Förster, T. Zwischenmolekulare energiewanderung und fluoreszenz. Ann. Phys. 1948, 437, 55–75. [Google Scholar] [CrossRef]
- Stryer, L.; Haugland, R.P. Energy transfer: A spectroscopic ruler. Proc. Natl. Acad. Sci. USA 1967, 58, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Latt, S.A.; Cheung, H.T.; Blout, E.R. Energy transfer: A System with relatively fixed donor-acceptor separation. J. Am. Chem. Soc. 1965, 87, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Stryer, L. Fluorescence energy transfer as a spectroscopic ruler. Annu. Rev. Biochem. 1978, 47, 819–846. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Vereb, G.; Post, J.N.; Friedländer, E.; Szöllösi, J. Novel single cell fluorescence approaches in the investigation of signaling at the cellular level. In Biophysical Aspects of Transmembrane Signaling; Damjanovich, S., Ed.; Springer-Verlag: Heidelberg, Germany, 2005; pp. 33–70. [Google Scholar]
- Szollosi, J.; Damjanovich, S.; Matyus, L. Application of fluorescence resonance energy transfer in the clinical laboratory: Routine and research. Cytometry 1998, 34, 159–179. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer Science & Business Media: New York, NY, USA, 2006. [Google Scholar]
- Wieb van der Meer, B. Förster theory. In FRET—Förster Resonance Energy Transfer:From Theory to Applications; Medintz, I., Hildebrant, N., Eds.; Wiley-VCH: Wienheim, Germany, 2014; pp. 23–62. [Google Scholar]
- Lidke, D.S.; Nagy, P.; Barisas, B.G.; Heintzmann, R.; Post, J.N.; Lidke, K.A.; Clayton, A.H.; Arndt-Jovin, D.J.; Jovin, T.M. Imaging molecular interactions in cells by dynamic and static fluorescence anisotropy (rFLIM and emFRET). Biochem. Soc. Trans. 2003, 31, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.T.; Kaminski, C.F.; Kaminski Schierle, G.S. HomoFRET Fluorescence Anisotropy Imaging as a tool to study molecular self-assembly in live cells. Chemphyschem 2011, 12, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Vereb, G.; Nagy, P.; Szöllösi, J. Flow cytometric FRET analysis of protein interaction. In Flow Cytometry Protocols, 3rd ed.; Hawley, T.S., Hawley, R.G., Eds.; Humana Press, Springer Science & Business Media: New York, NY, USA, 2011; pp. 371–392. [Google Scholar]
- Dale, R.E.; Eisinger, J.; Blumberg, W.E. The orientational freedom of molecular probes. The orientation factor in intramolecular energy transfer. Biophys. J. 1979, 26, 161–193. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer, B.W. κ-Squared: From nuisance to new sense. J. Biotechnol. 2002, 82, 181–196. [Google Scholar] [PubMed]
- Byrne, A.G.; Byrne, M.M.; Coker, G., III; Gemmill, K.B.; Spillmann, C.; Medintz, I.; Sloan, S.L.; Wieb van der Meer, B. Data. In FRET—Förster Resonance Energy Transfer: From Theory to Applications; Medintz, I., Hildebrant, N., Eds.; Wiley-VCH: Wienheim, Germany, 2014; pp. 657–755. [Google Scholar]
- Muller, S.M.; Galliardt, H.; Schneider, J.; Barisas, B.G.; Seidel, T. Quantification of Förster resonance energy transfer by monitoring sensitized emission in living plant cells. Front. Plant. Sci. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, D.; Bagosi, A.; Szollosi, J.; Jenei, A. Comparative study of the three different fluorophore antibody conjugation strategies. Anal. Bioanal. Chem. 2012, 404, 1449–1463. [Google Scholar] [CrossRef] [PubMed]
- Zal, T.; Gascoigne, N.R. Photobleaching-corrected FRET efficiency imaging of live cells. Biophys. J. 2004, 86, 3923–3939. [Google Scholar] [CrossRef] [PubMed]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. Engl. 2009, 48, 6974–6998. [Google Scholar] [CrossRef] [PubMed]
- Lofblom, J.; Feldwisch, J.; Tolmachev, V.; Carlsson, J.; Stahl, S.; Frejd, F.Y. Affibody molecules: Engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010, 584, 2670–2680. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, G.T. Bioconjugate Techniques; Academic Press: San Diego, CA, USA, 2013. [Google Scholar]
- Sapsford, K.E.; Berti, L.; Medintz, I.L. Materials for fluorescence resonance energy transfer analysis: Beyond traditional donor-acceptor combinations. Angew. Chem. Int. Ed. Engl. 2006, 45, 4562–4589. [Google Scholar] [CrossRef] [PubMed]
- Horvath, G.; Petras, M.; Szentesi, G.; Fabian, A.; Park, J.W.; Vereb, G.; Szollosi, J. Selecting the right fluorophores and flow cytometer for fluorescence resonance energy transfer measurements. Cytom. A 2005, 65, 148–157. [Google Scholar] [CrossRef]
- Sebestyen, Z.; Nagy, P.; Horvath, G.; Vamosi, G.; Debets, R.; Gratama, J.W.; Alexander, D.R.; Szollosi, J. Long wavelength fluorophores and cell-by-cell correction for autofluorescence significantly improves the accuracy of flow cytometric energy transfer measurements on a dual-laser benchtop flow cytometer. Cytometry 2002, 48, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, V.J.; Levisson, M.; Eppink, M.H.; Smidt, H.; van der Oost, J. Alternative affinity tools: More attractive than antibodies? Biochem. J. 2011, 436, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Miersch, S.; Sidhu, S.S. Synthetic antibodies: Concepts, potential and practical considerations. Methods 2012, 57, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed]
- Schnell, U.; Dijk, F.; Sjollema, K.A.; Giepmans, B.N. Immunolabeling artifacts and the need for live-cell imaging. Nat. Methods 2012, 9, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.; Chott, A.; Fabiano, A.; Battifora, H. Effect of formalin tissue fixation and processing on immunohistochemistry. Am. J. Surg. Pathol. 2000, 24, 1016–1019. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Piatkevich, K.D.; Lionnet, T.; Singer, R.H.; Verkhusha, V.V. Modern fluorescent proteins and imaging technologies to study gene expression, nuclear localization, and dynamics. Curr. Opin. Cell. Biol. 2011, 23, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, M. Introduction to fluorescent proteins. In The Fluorescent Protein Revolution; Periasamy, A., Ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2014; pp. 3–24. [Google Scholar]
- Day, R.N. Fluorescent proteins for FRET: Monitoring protein interactions in living cells. In The Fluorescent Protein Revolution; Periasamy, A., Ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2014; pp. 245–278. [Google Scholar]
- Dedecker, P.; de Schryver, F.C.; Hofkens, J. Fluorescent proteins: Shine on, you crazy diamond. J. Am. Chem. Soc. 2013, 135, 2387–2402. [Google Scholar] [CrossRef] [PubMed]
- Piston, D.W.; Kremers, G.J. Fluorescent protein FRET: The good, the bad and the ugly. Trends Biochem. Sci. 2007, 32, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.J.; St-Pierre, F.; Gong, Y.; Marshall, J.D.; Cranfill, P.J.; Baird, M.A.; McKeown, M.R.; Wiedenmann, J.; Davidson, M.W.; Schnitzer, M.J.; et al. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 2012, 9, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Marsh, D.R.; Holmes, K.D.; Dekaban, G.A.; Weaver, L.C. Distribution of an NMDA receptor:GFP fusion protein in sensory neurons is altered by a C-terminal construct. J. Neurochem. 2001, 77, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, D.; Okumus, B.; Chien, P.; Baker, T.A.; Paulsson, J. Segregation of molecules at cell division reveals native protein localization. Nat. Methods 2012, 9, U480–U498. [Google Scholar] [CrossRef]
- Ettinger, A.; Wittmann, T. Fluorescence live cell imaging. Methods Cell. Biol. 2013, 123, 77–94. [Google Scholar]
- Costantini, L.M.; Fossati, M.; Francolini, M.; Snapp, E.L. Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions. Traffic 2012, 13, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Crivat, G.; Taraska, J.W. Imaging proteins inside cells with fluorescent tags. Trends Biotechnol. 2012, 30, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.; Ting, A.Y. Site-specific labeling of proteins with small molecules in live cells. Curr. Opin. Biotechnol. 2005, 16, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Gautier, A.; Juillerat, A.; Heinis, C.; Correa, I.R., Jr.; Kindermann, M.; Beaufils, F.; Johnsson, K. An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 2008, 15, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Xu-Welliver, M.; Pegg, A.E. Degradation of the alkylated form of the DNA repair protein, O6-alkylguanine-DNA alkyltransferase. Carcinogenesis 2002, 23, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, C.P.; Vereb, G.; Geshi, N.; Schulz, A. Inhibition of cytoplasmic streaming by cytochalasin D is superior to paraformaldehyde Fixation for measuring FRET between fluorescent protein-tagged golgi components. Cytom. Part A 2013, 83, 830–838. [Google Scholar] [CrossRef]
- Epe, B.; Woolley, P.; Steinhauser, K.G.; Littlechild, J. Distance measurement by energy transfer: The 3' end of 16S RNA and proteins S4 and S17 of the ribosome of Escherichia coli. Eur. J. Biochem. 1982, 129, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Jovin, T.; Arndt-Jovin, D. FRET microscopy: Digital imaging of fluorescence resonance energy transfer. Application in cell biology. Cell. Struct. Funct. Microspectrofluorometry 1989, 30, 99–117. [Google Scholar]
- Szollosi, J.; Tron, L.; Damjanovich, S.; Helliwell, S.H.; Arndt-Jovin, D.; Jovin, T.M. Fluorescence energy transfer measurements on cell surfaces: A critical comparison of steady-state fluorimetric and flow cytometric methods. Cytometry 1984, 5, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.S.; Arndt-Jovin, D.J.; Jovin, T.M. Proximity of lectin receptors on the cell surface measured by fluorescence energy transfer in a flow system. J. Histochem. Cytochem. 1979, 27, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Zal, T.; Zal, M.A.; Gascoigne, N.R. Inhibition of T cell receptor-coreceptor interactions by antagonist ligands visualized by live FRET imaging of the T-Hybridoma immunological synapse. Immunity 2002, 16, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Zeug, A.; Woehler, A.; Neher, E.; Ponimaskin, E.G. Quantitative intensity-based FRET approaches—A comparative snapshot. Biophys. J. 2012, 103, 1821–1827. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G.W.; Berry, G.; Liang, X.H.; Levine, B.; Herman, B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 1998, 74, 2702–2713. [Google Scholar] [CrossRef] [PubMed]
- Szaloki, N.; Doan-Xuan, Q.M.; Szollosi, J.; Toth, K.; Vamosi, G.; Bacso, Z. High throughput FRET analysis of protein–protein interactions by slide-based imaging laser scanning cytometry. Cytom. A 2013, 83, 818–829. [Google Scholar] [CrossRef] [Green Version]
- Mittag, A.; Lenz, D.; Bocsi, J.; Sack, U.; Gerstner, A.O.; Tarnok, A. Sequential photobleaching of fluorochromes for polychromatic slide-based cytometry. Cytom. A 2006, 69, 139–141. [Google Scholar] [CrossRef]
- Szabo, G., Jr.; Pine, P.S.; Weaver, J.L.; Kasari, M.; Aszalos, A. Epitope mapping by photobleaching fluorescence resonance energy transfer measurements using a laser scanning microscope system. Biophys. J. 1992, 61, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Jares-Erijman, E.A.; Jovin, T.M. FRET imaging. Nat. Biotechnol. 2003, 21, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Berney, C.; Danuser, G. FRET or no FRET: A quantitative comparison. Biophys. J. 2003, 84, 3992–4010. [Google Scholar] [CrossRef] [PubMed]
- Szöllösi, J.; Damjanovich, S.; Nagy, P.; Vereb, G.; Mátyus, L. Principles of resonance energy transfer. Curr. Protoc. Cytom. 2006. [Google Scholar] [CrossRef]
- Zal, T.; Gascoigne, N.R. Using live FRET imaging to reveal early protein–protein interactions during T cell activation. Curr. Opin. Immunol. 2004, 16, 674–683. [Google Scholar] [PubMed]
- Ishikawa-Ankerhold, H.C.; Ankerhold, R.; Drummen, G.P. Advanced fluorescence microscopy techniques—FRAP, FLIP, FLAP, FRET and FLIM. Molecules 2012, 17, 4047–4132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, P.; Szabo, A.; Varadi, T.; Kovacs, T.; Batta, G.; Szollosi, J. Maximum likelihood estimation of FRET efficiency and its implications for distortions in pixelwise calculation of FRET in microscopy. Cytom. A 2014, 85, 942–952. [Google Scholar] [CrossRef]
- Szabo, A.; Szollosi, J.; Nagy, P. Coclustering of ErbB1 and ErbB2 revealed by FRET-sensitized acceptor bleaching. Biophys. J. 2010, 99, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Bene, L.; Hyun, W.C.; Vereb, G.; Braun, M.; Antz, C.; Paysan, J.; Damjanovich, S.; Park, J.W.; Szollsi, J. Novel calibration method for flow cytometric fluorescence resonance energy transfer measurements between visible fluorescent proteins. Cytom. A 2005, 67, 86–96. [Google Scholar] [CrossRef]
- Van Wageningen, S.; Pennings, A.H.; van der Reijden, B.A.; Boezeman, J.B.; de Lange, F.; Jansen, J.H. Isolation of FRET-positive cells using single 408-nm laser flow cytometry. Cytom. A 2006, 69, 291–298. [Google Scholar] [CrossRef]
- Paar, C.; Paster, W.; Stockinger, H.; Schutz, G.J.; Sonnleitner, M.; Sonnleitner, A. High throughput FRET screening of the plasma membrane based on TIRFM. Cytom. A 2008, 73, 442–450. [Google Scholar] [CrossRef]
- Nagy, P.; Vereb, G.; Damjanovich, S.; Mátyus, L.; Szöllösi, J. Measuring FRET in flow cytometry and microscopy. Curr. Protoc. Cytom. 2006. [Google Scholar] [CrossRef]
- Nagy, P.; Vamosi, G.; Bodnar, A.; Lockett, S.J.; Szollosi, J. Intensity-based energy transfer measurements in digital imaging microscopy. Eur. Biophys. J. 1998, 27, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Roszik, J.; Lisboa, D.; Szollosi, J.; Vereb, G. Evaluation of intensity-based ratiometric FRET in image cytometry-approaches and a software solution. Cytom. A 2009, 75, 761–767. [Google Scholar] [CrossRef]
- Roszik, J.; Szollosi, J.; Vereb, G. AccPbFRET: An ImageJ plugin for semi-automatic, fully corrected analysis of acceptor photobleaching FRET images. BMC Bioinform. 2008, 9. [Google Scholar] [CrossRef]
- Giordano, L.; Jovin, T.M.; Irie, M.; Jares-Erijman, E.A. Diheteroarylethenes as thermally stable photoswitchable acceptors in photochromic fluorescence resonance energy transfer (pcFRET). J. Am. Chem. Soc. 2002, 124, 7481–7489. [Google Scholar] [CrossRef] [PubMed]
- Subach, F.V.; Zhang, L.; Gadella, T.W.; Gurskaya, N.G.; Lukyanov, K.A.; Verkhusha, V.V. Red fluorescent protein with reversibly photoswitchable absorbance for photochromic FRET. Chem. Biol. 2010, 17, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Vanderklish, P.W.; Krushel, L.A.; Holst, B.H.; Gally, J.A.; Crossin, K.L.; Edelman, G.M. Marking synaptic activity in dendritic spines with a calpain substrate exhibiting fluorescence resonance energy transfer. Proc. Natl. Acad. Sci USA 2000, 97, 2253–2258. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.W.; Daugherty, P.S. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat. Biotechnol. 2005, 23, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Larbret, F.; Dubois, N.; Brau, F.; Guillemot, E.; Mahiddine, K.; Tartare-Deckert, S.; Verhasselt, V.; Deckert, M. Technical advance: Actin CytoFRET, a novel FRET flow cytometry method for detection of actin dynamics in resting and activated T cell. J. Leukoc. Biol. 2013, 94, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Jalink, K.; van Rheenen, J. FilterFRET: Quantitative imaging of sensitized emission. In FRET and FLIM Techniques; Gadella, T., Ed.; Elsevier: Amsterdam, The Netherlands, 2009; Volume 33, pp. 289–349. [Google Scholar]
- Adams, S.R.; Harootunian, A.T.; Buechler, Y.J.; Taylor, S.S.; Tsien, R.Y. Fluorescence ratio imaging of cyclic AMP in single cells. Nature 1991, 349, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Youvan, D.C.; Silva, C.M.; Bylina, E.J.; Coleman, W.J.; Dilworth, M.R.; Yang, M.M. Calibration of fluorescence resonance energy transfer in microscopy using genetically engineered GFP derivatives on nickel chelating beads. Biotechnology 1997, 3, 1–18. [Google Scholar]
- Erickson, M.G.; Alseikhan, B.A.; Peterson, B.Z.; Yue, D.T. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron 2001, 31, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Szollosi, J.; Nagy, P.; Sebestyen, Z.; Damjanovicha, S.; Park, J.W.; Matyus, L. Applications of fluorescence resonance energy transfer for mapping biological membranes. J. Biotechnol. 2002, 82, 251–266. [Google Scholar] [PubMed]
- Chen, H.; Puhl, H.L., 3rd; Koushik, S.V.; Vogel, S.S.; Ikeda, S.R. Measurement of FRET efficiency and ratio of donor to acceptor concentration in living cells. Biophys. J. 2006, 91, L39–L41. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Liu, Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys. J. 2001, 81, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, A.; Christensen, K.; Swanson, J.A. Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys. J. 2002, 83, 3652–3664. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Sorkin, A. Coordinated traffic of Grb2 and Ras during epidermal growth factor receptor endocytosis visualized in living cells. Mol. Biol. Cell. 2002, 13, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Bene, L.; Ungvari, T.; Fedor, R.; Sasi Szabo, L.; Damjanovich, L. Intensity correlation-based calibration of FRET. Biophys. J. 2013, 105, 2024–2035. [Google Scholar] [CrossRef] [PubMed]
- Vamosi, G.; Baudendistel, N.; von der Lieth, C.W.; Szaloki, N.; Mocsar, G.; Muller, G.; Brazda, P.; Waldeck, W.; Damjanovich, S.; Langowski, J.; et al. Conformation of the c-Fos/c-Jun complex in vivo: A combined FRET, FCCS, and MD-modeling study. Biophys. J. 2008, 94, 2859–2868. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-I. Hyperspectral Imaging: Techniques for Spectral Detection and Classification; Springer Science & Business Media: Berlin, Germany, 2003; Volume 1. [Google Scholar]
- Zimmermann, T.; Rietdorf, J.; Girod, A.; Georget, V.; Pepperkok, R. Spectral imaging and linear un-mixing enables improved FRET efficiency with a novel GFP2-YFP FRET pair. FEBS Lett. 2002, 531, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mauldin, J.P.; Day, R.N.; Periasamy, A. Characterization of spectral FRET imaging microscopy for monitoring nuclear protein interactions. J. Microsc. 2007, 228, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Megias, D.; Marrero, R.; Martinez Del Peso, B.; Garcia, M.A.; Bravo-Cordero, J.J.; Garcia-Grande, A.; Santos, A.; Montoya, M.C. Novel lambda FRET spectral confocal microscopy imaging method. Microsc. Res. Technol. 2009, 72, 1–11. [Google Scholar] [CrossRef]
- Levy, S.; Wilms, C.D.; Brumer, E.; Kahn, J.; Pnueli, L.; Arava, Y.; Eilers, J.; Gitler, D. SpRET: Highly sensitive and reliable spectral measurement of absolute fret efficiency. Microsc. Microanal. 2011, 17, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Broussard, J.A.; Rappaz, B.; Webb, D.J.; Brown, C.M. Fluorescence resonance energy transfer microscopy as demonstrated by measuring the activation of the serine/threonine kinase Akt. Nat. Protoc. 2013, 8, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Leavesley, S.J.; Britain, A.L.; Cichon, L.K.; Nikolaev, V.O.; Rich, T.C. Assessing FRET using spectral techniques. Cytom. Part A 2013, 83, 898–912. [Google Scholar]
- Kim, J.; Li, X.; Kang, M.S.; Im, K.B.; Genovesio, A.; Grailhe, R. Quantification of protein interaction in living cells by two-photon spectral imaging with fluorescent protein fluorescence resonance energy transfer pair devoid of acceptor bleed-through. Cytom. A 2012, 81, 112–119. [Google Scholar] [CrossRef]
- Thaler, C.; Koushik, S.V.; Blank, P.S.; Vogel, S.S. Quantitative multiphoton spectral imaging and its use for measuring resonance energy transfer. Biophys. J. 2005, 89, 2736–2749. [Google Scholar] [CrossRef] [PubMed]
- Szentesi, G.; Vereb, G.; Horvath, G.; Bodnar, A.; Fabian, A.; Matko, J.; Gaspar, R.; Damjanovich, S.; Matyus, L.; Jenei, A. Computer program for analyzing donor photobleaching FRET image series. Cytom. A 2005, 67, 119–128. [Google Scholar] [CrossRef]
- Clayton, A.H.; Klonis, N.; Cody, S.H.; Nice, E.C. Dual-channel photobleaching FRET microscopy for improved resolution of protein association states in living cells. Eur. Biophys. J. 2005, 34, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Varma, C.A.; Verhoeven, J.W.; Tanke, H.J. Influence of the triplet excited state on the photobleaching kinetics of fluorescein in microscopy. Biophys. J. 1996, 70, 2959–2968. [Google Scholar] [CrossRef] [PubMed]
- Redford, G.; Clegg, R.M. Real-time fluorescence lifetime imaging and FRET using fast gated image intensifiers. In Molecular Imaging: FRET Microscopy and Spectroscopy; Periasamy, A., Day, R.D., Eds.; Oxford University Press: New York, NY, USA, 2005; pp. 193–226. [Google Scholar]
- Berezin, M.Y.; Achilefu, S. Fluorescence lifetime measurements and biological imaging. Chem. Rev. 2010, 110, 2641–2684. [Google Scholar] [CrossRef] [PubMed]
- Wouters, F.S. Förster resonance energy transfer and fluorescence lifetime imaging. Fluoresc. Microsc. 2013, 245–291. [Google Scholar]
- Szabo, A.; Horvath, G.; Szollosi, J.; Nagy, P. Quantitative characterization of the large-scale association of ErbB1 and ErbB2 by flow cytometric homo-FRET measurements. Biophys. J. 2008, 95, 2086–2096. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.S.; Thaler, C.; Blank, P.S.; Koushik, S.V. Time resolved fluorescence anisotropy. FLIM Microsc. Biol. Med. 2009, 1, 245–288. [Google Scholar]
- LiCata, V.J.; Wowor, A.J. Applications of fluorescence anisotropy to the study of protein-DNA interactions. Methods Cell. Biol. 2008, 84, 243–262. [Google Scholar] [PubMed]
- Fernandez, S.M.; Berlin, R.D. Cell surface distribution of lectin receptors determined by resonance energy transfer. Nature 1976, 264, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, G.L. The fluid-mosaic model of membrane structure: Still relevant to understanding the structure, function and dynamics of biological membranes after more than 40 years. Biochim. Biophys. Acta 2014, 1838, 1451–1466. [Google Scholar] [CrossRef] [PubMed]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- IUIS-WHO Nomenclature Subcommittee. Nomenclature for clusters of differentiation (CD) of antigens defined on human leukocyte populations. Bull. World Health Organ. 1984, 62, 809–815. [Google Scholar]
- Vereb, G.; Szollosi, J.; Matko, J.; Nagy, P.; Farkas, T.; Vigh, L.; Matyus, L.; Waldmann, T.A.; Damjanovich, S. Dynamic, yet structured: The cell membrane three decades after the singer-nicolson model. Proc. Natl. Acad. Sci. USA 2003, 100, 8053–8058. [Google Scholar] [CrossRef] [PubMed]
- Layre, E.; de Jong, A.; Moody, D.B. Human T cells use CD1 and MR1 to recognize lipids and small molecules. Curr. Opin. Chem. Biol. 2014, 23C, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Neppert, J.; Mueller-Eckhardt, C. Monoclonal mouse antibodies to human MHC class I antigens cocap class II antigens. Tissue Antigens 1984, 24, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Szollosi, J.; Damjanovich, S.; Balazs, M.; Nagy, P.; Tron, L.; Fulwyler, M.J.; Brodsky, F.M. Physical association between MHC class I and class II molecules detected on the cell surface by flow cytometric energy transfer. J. Immunol. 1989, 143, 208–213. [Google Scholar] [PubMed]
- Szollosi, J.; Horejsi, V.; Bene, L.; Angelisova, P.; Damjanovich, S. Supramolecular complexes of MHC class I, MHC class II, CD20, and tetraspan molecules (CD53, CD81, and CD82) at the surface of a B cell line JY. J. Immunol. 1996, 157, 2939–2946. [Google Scholar] [PubMed]
- Shrestha, D.; Exley, M.A.; Vereb, G.; Szollosi, J.; Jenei, A. CD1d favors MHC neighborhood, GM1 ganglioside proximity and low detergent sensitive membrane regions on the surface of B lymphocytes. Biochim. Biophys. Acta 2014, 1840, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Baumgarth, N.; Dick, M.D.; Brossay, L.; Kronenberg, M.; Herzenberg, L.A.; Strober, S. CD1 expression defines subsets of follicular and marginal zone B cells in the spleen: β2-Microglobulin-dependent and independent forms. J. Immunol. 1998, 161, 1710–1717. [Google Scholar] [PubMed]
- Gourapura, R.J.; Khan, M.A.; Gallo, R.M.; Shaji, D.; Liu, J.; Brutkiewicz, R.R. Forming a complex with MHC class I molecules interferes with mouse CD1d functional expression. PLoS ONE 2013, 8, e72867. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, X.; Keaton, J.M.; Reddington, F.; Illarionov, P.A.; Besra, G.S.; Gumperz, J.E. Distinct endosomal trafficking requirements for presentation of autoantigens and exogenous lipids by human CD1d molecules. J. Immunol. 2007, 178, 6181–6190. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.D.; Moseley, G.W.; van Spriel, A.B. Tetraspanin microdomains in immune cell signalling and malignant disease. Tissue Antigens 2004, 64, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Sloma, I.; Zilber, M.T.; Vasselon, T.; Setterblad, N.; Cavallari, M.; Mori, L.; de Libero, G.; Charron, D.; Mooney, N.; Gelin, C. Regulation of CD1a Surface expression and antigen presentation by invariant chain and lipid rafts. J. Immunol. 2008, 180, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.J.; Cresswell, P. Regulation of intracellular trafficking of human CD1d by association with MHC class II molecules. EMBO J. 2002, 21, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Van den Hoorn, T.; Paul, P.; Janssen, L.; Janssen, H.; Neefjes, J. Dynamics within tetraspanin pairs affect MHC class II expression. J. Cell Sci. 2012, 125, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Bene, L.; Kanyari, Z.; Bodnar, A.; Kappelmayer, J.; Waldmann, T.A.; Vamosi, G.; Damjanovich, L. Colorectal carcinoma rearranges cell surface protein topology and density in CD4+ T cells. Biochem. Biophys. Res. Commun. 2007, 361, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Bene, L.; Balazs, M.; Matko, J.; Most, J.; Dierich, M.P.; Szollosi, J.; Damjanovich, S. Lateral organization of the ICAM-1 molecule at the surface of human lymphoblasts: A possible model for its co-distribution with the IL-2 receptor, class I and class II HLA molecules. Eur. J. Immunol. 1994, 24, 2115–2123. [Google Scholar] [CrossRef] [PubMed]
- Amiot, M.; Dastot, H.; Fabbi, M.; Degos, L.; Bernard, A.; Boumsell, L. Intermolecular complexes between three human CD1 molecules on normal thymus cells. Immunogenetics 1988, 27, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Amiot, M.; Dastot, H.; Degos, L.; Dausset, J.; Bernard, A.; Boumsell, L. HLA class I molecules are associated with CD1a heavy chains on normal human thymus cells. Proc. Natl. Acad. Sci. USA 1988, 85, 4451–4454. [Google Scholar] [CrossRef] [PubMed]
- Amiot, M.; Dastot, H.; Schmid, M.; Bernard, A.; Boumsell, L. Analysis of CD1 molecules on thymus cells and leukemic T lymphoblasts identifies discrete phenotypes and reveals that CD1 intermolecular complexes are observed only on normal cells. Blood 1987, 70, 676–685. [Google Scholar] [PubMed]
- Bodnar, A.; Nizsaloczki, E.; Mocsar, G.; Szaloki, N.; Waldmann, T.A.; Damjanovich, S.; Vamosi, G. A biophysical approach to IL-2 and IL-15 receptor function: Localization, conformation and interactions. Immunol. Lett. 2008, 116, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Vamosi, G.; Bodnar, A.; Vereb, G.; Jenei, A.; Goldman, C.K.; Langowski, J.; Toth, K.; Matyus, L.; Szollosi, J.; Waldmann, T.A.; et al. IL-2 and IL-15 receptor α-subunits are coexpressed in a supramolecular receptor cluster in lipid rafts of T cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11082–11087. [Google Scholar] [CrossRef] [PubMed]
- Damjanovich, S.; Bene, L.; Matko, J.; Alileche, A.; Goldman, C.K.; Sharrow, S.; Waldmann, T.A. Preassembly of interleukin 2 (IL-2) receptor subunits on resting Kit 225 K6 T cells and their modulation by IL-2, IL-7, and IL-15: A fluorescence resonance energy transfer study. Proc. Natl. Acad. Sci. USA 1997, 94, 13134–13139. [Google Scholar] [CrossRef] [PubMed]
- Damjanovich, L.; Volko, J.; Forgacs, A.; Hohenberger, W.; Bene, L. Crohn’s Disease Alters MHC-rafts in CD4+ T-cells. Cytom. A 2012, 81, 149–164. [Google Scholar] [CrossRef]
- De Menthon, M.; Lambert, M.; Guiard, E.; Tognarelli, S.; Bienvenu, B.; Karras, A.; Guillevin, L.; Caillat-Zucman, S. Excessive interleukin-15 transpresentation endows NKG2D+CD4+ T cells with innate-like capacity to lyse cascular endothelium in granulomatosis with polyangiitis (Wegener’s). Arthritis Rheumatol. 2011, 63, 2116–2126. [Google Scholar] [CrossRef]
- Huppa, J.B.; Davis, M.M. T-cell-antigen recognition and the immunological synapse. Nat. Rev. Immunol. 2003, 3, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R.S.; Goldman, S.J.; Spitalny, G.L.; Burakoff, S.J. T-cell receptor-CD4 physical association in a murine T-cell hybridoma: Induction by antigen receptor ligation. Proc. Natl. Acad. Sci. USA 1989, 86, 8531–8535. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.L.; Uniyal, S.; Shin, J.; Strominger, J.L.; Mittler, R.; Burakoff, S. p56lck Association with CD4 is required for the interaction between CD4 and the TCR/CD3 complex and for optimal antigen stimulation. J. Immunol. 1992, 148, 2159–2162. [Google Scholar] [PubMed]
- Bacso, Z.; Bene, L.; Bodnar, A.; Matko, J.; Damjanovich, S. A photobleaching energy transfer analysis of CD8/MHC-I and LFA-1/ICAM-1 interactions in CTL-target cell conjugates. Immunol. Lett. 1996, 54, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Yachi, P.P.; Ampudia, J.; Gascoigne, N.R.; Zal, T. Nonstimulatory peptides contribute to antigen-induced CD8-T cell receptor interaction at the immunological synapse. Nat. Immunol. 2005, 6, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Roszik, J.; Sebestyen, Z.; Govers, C.; Guri, Y.; Szoor, A.; Palyi-Krekk, Z.; Vereb, G.; Nagy, P.; Szollosi, J.; Debets, R. T-cell synapse formation depends on antigen recognition but not CD3 interaction: Studies with TCR: ζ, a candidate transgene for TCR gene therapy. Eur. J. Immunol. 2011, 41, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Dornan, S.; Sebestyen, Z.; Gamble, J.; Nagy, P.; Bodnar, A.; Alldridge, L.; Doe, S.; Holmes, N.; Goff, L.K.; Beverley, P.; et al. Differential association of CD45 isoforms with CD4 and CD8 regulates the actions of specific pools of p56lck tyrosine kinase in T cell antigen receptor signal transduction. J. Biol. Chem. 2002, 277, 1912–1918. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.H.; Maruyama, I.N. All EGF (ErbB) receptors have preformed homo- and heterodimeric structures in living cells. J. Cell Sci. 2008, 121, 3207–3217. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Vereb, G.; Sebestyen, Z.; Horvath, G.; Lockett, S.J.; Damjanovich, S.; Park, J.W.; Jovin, T.M.; Szollosi, J. Lipid rafts and the local density of ErbB proteins influence the biological role of homo- and heteroassociations of ErbB2. J. Cell Sci. 2002, 115, 4251–4262. [Google Scholar] [CrossRef] [PubMed]
- Fazekas, Z.; Petras, M.; Fabian, A.; Palyi-Krekk, Z.; Nagy, P.; Damjanovich, S.; Vereb, G.; Szollosi, J. Two-sided fluorescence resonance energy transfer for assessing molecular interactions of up to three distinct species in confocal microscopy. Cytom. A 2008, 73, 209–219. [Google Scholar] [CrossRef]
- Petras, M.; Lajtos, T.; Friedlander, E.; Klekner, A.; Pintye, E.; Feuerstein, B.G.; Szollosi, J.; Vereb, G. Molecular interactions of ErbB1 (EGFR) and integrin-β1 in astrocytoma frozen sections predict clinical outcome and correlate with Akt-mediated in vitro radioresistance. Neuro Oncol. 2013, 15, 1027–1040. [Google Scholar] [CrossRef] [PubMed]
- Maliwal, B.P.; Raut, S.; Fudala, R.; D’Auria, S.; Marzullo, V.M.; Luini, A.; Gryczynski, I.; Gryczynski, Z. Extending Förster resonance energy transfer measurements beyond 100 å using common organic fluorophores: Enhanced transfer in the presence of multiple acceptors. J. Biomed. Opt. 2012, 17, 0110061–0110068. [Google Scholar] [CrossRef]
- Yun, C.S.; Javier, A.; Jennings, T.; Fisher, M.; Hira, S.; Peterson, S.; Hopkins, B.; Reich, N.O.; Strouse, G.F. Nanometal surface energy transfer in optical rulers, breaking the FRET barrier. J. Am. Chem. Soc. 2005, 127, 3115–3119. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.S.; van der Meer, B.W.; Blank, P.S. Estimating the distance separating fluorescent protein FRET pairs. Methods 2014, 66, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Fabian, A.; Horvath, G.; Vamosi, G.; Vereb, G.; Szollosi, J. TripleFRET measurements in flow cytometry. Cytom. A 2013, 83, 375–385. [Google Scholar] [CrossRef]
- Koushik, S.V.; Blank, P.S.; Vogel, S.S. Anomalous surplus energy transfer observed with multiple FRET acceptors. PLoS ONE 2009, 4, e8031. [Google Scholar] [CrossRef] [PubMed]
- Schuler, B. Single-molecule FRET of protein structure and dynamics—A primer. J. Nanobiotechnol. 2013, 11 (Suppl. 1). [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shrestha, D.; Jenei, A.; Nagy, P.; Vereb, G.; Szöllősi, J. Understanding FRET as a Research Tool for Cellular Studies. Int. J. Mol. Sci. 2015, 16, 6718-6756. https://doi.org/10.3390/ijms16046718
Shrestha D, Jenei A, Nagy P, Vereb G, Szöllősi J. Understanding FRET as a Research Tool for Cellular Studies. International Journal of Molecular Sciences. 2015; 16(4):6718-6756. https://doi.org/10.3390/ijms16046718
Chicago/Turabian StyleShrestha, Dilip, Attila Jenei, Péter Nagy, György Vereb, and János Szöllősi. 2015. "Understanding FRET as a Research Tool for Cellular Studies" International Journal of Molecular Sciences 16, no. 4: 6718-6756. https://doi.org/10.3390/ijms16046718
APA StyleShrestha, D., Jenei, A., Nagy, P., Vereb, G., & Szöllősi, J. (2015). Understanding FRET as a Research Tool for Cellular Studies. International Journal of Molecular Sciences, 16(4), 6718-6756. https://doi.org/10.3390/ijms16046718