
Modulation of the Genome and Epigenome of Individuals Susceptible to Autism by Environmental Risk Factors

Abstract
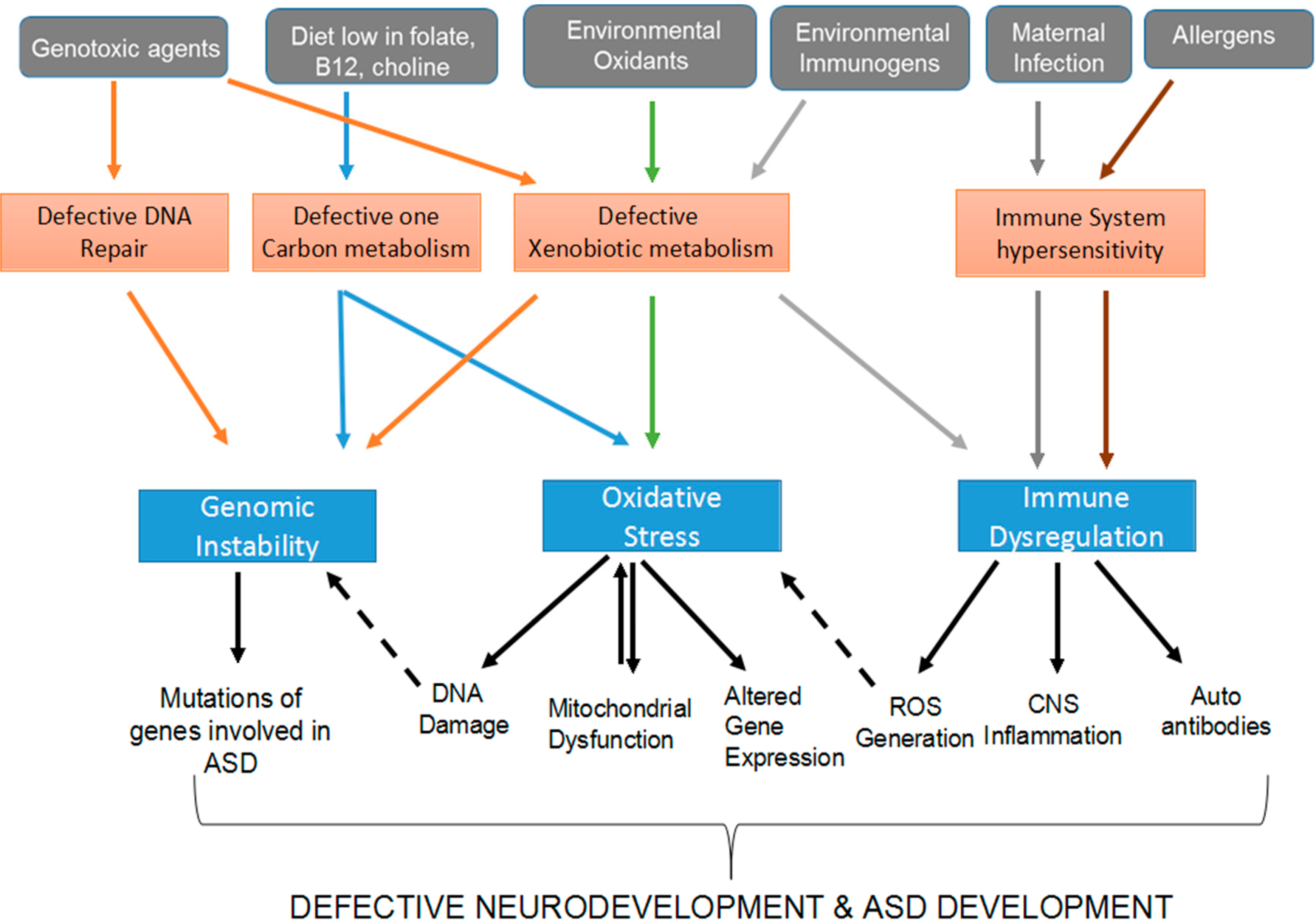
:1. Introduction
2. Disruption of the Genome and Epigenome of Genetically Susceptible Individuals by ASD Environmental Risk Factors
2.1. Defective Xenobiotic Metabolism of ASD Environmental Risk Factors
2.2. Increased Sensitivity to Endocrine-Disrupting Chemicals
2.3. Increased Susceptibility to Agents that Induce Oxidative Stress
2.4. Susceptibility to Epigenomic Dysregulation
2.5. Hyperactive Transposable Elements
2.6. Increased Genomic Instability
2.7. Abnormal Immune Activation
2.8. Gut Microbiota
2.9. Transgenerational Environmental Effects
2.10. Exacerbation of Environmental Effects by Seizures
3. Conclusions

{kind=link}
| Mechanisms | Effects on Genome and Epigenome | Environmental Factors | Genetic Factors |
|---|---|---|---|
| Oxidative stress | DNA damage; Mitochondria dysfunction; Disruption of the epigenome; Deregulated gene expression | Pro-oxidant toxicants; Diet low in one-carbon donors; Immunogens | Defective one-carbon metabolism; Defective anti-oxidant defenses; Abnormal immune activation; mitochondrial dysfunction |
| Abnormal immune activation | Autoantibody generation; CNS inflammation; Cytokine secretion; Gut permeability | Infections; Allergens; Immunogens | Defective xenobiotic metabolism; Autoimmune diseases; Hypersensitivity to immunogens |
| Genomic instability | Increased incidence of mutations predisposing to autism | DNA damaging agents; Diet low in one-carbon donors | Defective DNA repair; Defective xenobiotic metabolism; Genomic architecture |
| Epigenome dysregulation | Loss of normal gene regulation; Genomic instability | Heavy metals and toxins; CNS inflammation; Diet low in one-carbon donors | Defective one-carbon metabolism; Genomic architecture |
| Altered gut microbiome | Gut permeability; Neurotransmitter release; Immune dysfunction | Infections; Diet composition | Interaction of immune system with microbiota |
| Hyperactive transposable elements | Mutations affecting genes implicated in ASD | Chemicals activating retrotransposition | Reduced Mecp2 activity |
| Transgenerational inheritance | Abnormal gene expression patterns during neurodevelopment in unexposed generations | Perinatal stress; Endocrine disruptors; Effects on gut microbiome | Mechanisms are currently controversial |

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Geschwind, D.H. Advances in autism. Annu. Rev. Med. 2009, 60, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Devlin, B.; Scherer, S.W. Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 2012, 22, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; Giuliano, F.; Stordeur, C.; Depienne, C.; Mouzat, K.; et al. Meta-analysis of SHANK mutations in autism spectrum disorders: A gradient of severity in cognitive impairments. PLoS Genet. 2014, 10, e1004580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clipperton-Allen, A.E.; Page, D.T. Pten haploinsufficient mice show broad brain overgrowth but selective impairments in autism-relevant behavioral tests. Hum. Mol. Genet. 2014, 23, 3490–3505. [Google Scholar] [CrossRef] [PubMed]
- Tsai, N.P.; Wilkerson, J.R.; Guo, W.; Maksimova, M.A.; DeMartino, G.N.; Cowan, C.W.; Huber, K.M. Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell 2012, 151, 1581–1594. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, G.M.; Katz, D.M. Synaptic microcircuit dysfunction in genetic models of neurodevelopmental disorders: Focus on Mecp2 and Met. Curr. Opin. Neurobiol. 2011, 21, 827–833. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Shpyleva, S.; Melnyk, S.; Pavliv, O.; Pogribny, I.P. Elevated 5-hydroxymethylcytosine in the Engrailed-2 (EN-2) promoter is associated with increased gene expression and decreased MeCP2 binding in autism cerebellum. Transl. Psychiatry 2014, 4, e460. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; Rauch, T.A.; Pfeifer, G.P.; Hu, V.W. Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J. 2010, 24, 3036–3051. [Google Scholar] [CrossRef] [PubMed]
- Ladd-Acosta, C.; Hansen, K.D.; Briem, E.; Fallin, M.D.; Kaufmann, W.E.; Feinberg, A.P. Common DNA methylation alterations in multiple brain regions in autism. Mol. Psychiatry 2014, 19, 862–871. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Shpyleva, S.; Melnyk, S.; Pavliv, O.; Pogribny, I.P. Complex epigenetic regulation of the Engrailed-2 (EN-2) homeobox gene in the autism cerebellum. Transl. Psychiatry 2013, 3, e232. [Google Scholar] [CrossRef] [PubMed]
- Atladóttir, H.O.; Thorsen, P.; Østergaard, L.; Schendel, D.E.; Lemcke, S.; Abdallah, M.; Parner, E.T. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J. Autism Dev. Disord. 2010, 40, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.L.; Lyall, K.; Hart, J.E.; Laden, F.; Just, A.C.; Bobb, J.F.; Koenen, K.C.; Ascherio, A.; Weisskopf, M.G. Perinatal air pollutant exposures and autism spectrum disorder in the children of Nurses' Health Study II participants. Environ. Health Perspect. 2013, 121, 978–984. [Google Scholar] [PubMed]
- Surén, P.; Roth, C.; Bresnahan, M.; Haugen, M.; Hornig, M.; Hirtz, D.; Lie, K.K.; Lipkin, W.I.; Magnus, P.; Reichborn-Kjennerud, T.; et al. Association between maternal use of folic acid supplements and autism risk in children. JAMA 2013, 309, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Herbert, M.R. Contributions of the environment and environmentally vulnerable physiology to autism spectrum disorders. Curr. Opin. Neurol. 2010, 23, 103–110. [Google Scholar] [CrossRef] [PubMed]
- LaSalle, J.M. A genomic-point-of-view on environmental factors influencing the human brain methylome. Epigenetics 2011, 6, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Chaste, P.; Leboyer, M. Autism risk factors: Genes, environment, and gene-environment interactions. Dialogues Clin. Neurosci. 2012, 14, 281–292. [Google Scholar] [PubMed]
- LaSalle, J.M. Epigenomic strategies at the interface of genetic and environmental risk factors for autism. J. Hum. Genet. 2013, 58, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Stamou, M.; Streifel, K.M.; Goines, P.E.; Lein, P.J. Neuronal connectivity as a convergent target of gene × environment interactions that confer risk for Autism Spectrum Disorders. Neurotoxicol. Teratol. 2013, 36, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Miksys, S.; Tyndale, R.F. Cytochrome P450-mediated drug metabolism in the brain. J. Psychiatry Neurosci. 2013, 38, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Woods, R.; Vallero, R.O.; Golub, M.S.; Suarez, J.K.; Ta, T.A.; Yasui, D.H.; Chi, L.-H.; Kostyniak, P.J.; Pessah, I.N.; Berman, R.F.; et al. Long lived epigenetic interactions between perinatal PBDE exposure and Mecp22308 mutation. Hum. Mol. Genet. 2012, 21, 2399–2411. [Google Scholar] [CrossRef] [PubMed]
- Feo, M.L.; Gross, M.S.; McGarrigle, B.P.; Eljarrat, E.; Barceló, D.; Aga, D.S.; Olson, J.R. Biotransformation of BDE-47 to potentially toxic metabolites is predominantly mediated by human CYP2B6. Environ. Health Perspect. 2013, 121, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Kinney, D.K.; Barch, D.H.; Chayka, B.; Napoleon, S.; Munir, K.M. Environmental risk factors for autism: Do they help cause de novo genetic mutations? Med. Hypotheses 2010, 74, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Turesky, R.J. The role of genetic polymorphisms in the metabolism of carcinogenic heterocyclic amines. Curr. Drug Metab. 2004, 5, 169–180. [Google Scholar] [CrossRef] [PubMed]
- D’Amelio, M.; Ricci, I.; Sacco, R.; Liu, X.; D’Agruma, L.; Muscarella, L.A.; Guarnieri, V.; Militerni, R.; Bravaccio, C.; Elia, M.; et al. Paraoxonase gene variants are associated with autism in North America, but not in Italy: Possible regional specificity in gene-environment interactions. Mol. Psychiatry 2005, 10, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Eskenazi, B.; Huen, K.; Marks, A.; Harley, K.G.; Bradman, A.; Barr, D.B.; Holland, N. PON1 and neurodevelopment in children from the CHAMACOS study exposed to organophosphate pesticides in utero. Environ. Health Perspect. 2010, 118, 1775–1781. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Genuis, S.J.; Frye, R.E. Environmental toxicants and autism spectrum disorders: A systematic review. Transl. Psychiatry 2014, 4, e360. [Google Scholar] [CrossRef] [PubMed]
- Serajee, F.J.; Nabi, R.; Zhong, H.; Huq, M. Polymorphisms in xenobiotic metabolism genes and autism. J. Child Neurol. 2004, 19, 413–417. [Google Scholar] [PubMed]
- Buyske, S.; Williams, T.A.; Mars, A.E.; Stenroos, E.S.; Ming, S.X.; Wang, R.; Sreenath, M.; Factura, M.F.; Reddy, C.; Lambert, G.H.; et al. Analysis of case-parent trios at a locus with a deletion allele: Association of GSTM1 with autism. BMC Genet. 2006, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Baron-Cohen, S.; Knickmeyer, R.C.; Belmonte, M.K. Sex differences in the brain: Implications for explaining autism. Science 2005, 310, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Werling, D.M.; Geschwind, D.H. Sex differences in autism spectrum disorders. Curr. Opin. Neurol. 2013, 26, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, M.V.; Ashwin, E.; Auyeung, B.; Chakrabarti, B.; Taylor, K.; Hackett, G.; Bullmore, E.T.; Baron-Cohen, S. Fetal testosterone influences sexually dimorphic grey matter in the brain. J. Neurosci. 2012, 32, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Baron-Cohen, S.; Auyeung, B.; Nørgaard-Pedersen, B.; Hougaard, D.M.; Abdallah, M.W.; Melgaard, L.; Cohen, A.S.; Chakrabarti, B.; Ruta, L.; Lombardo, M.V. Elevated fetal steroidogenic activity in autism. Mol. Psychiatry 2015, 20, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Kajta, M.; Wójtowicz, A.K. Impact of endocrine-disrupting chemicals on neural development and the onset of neurological disorders. Pharmacol. Rep. 2013, 65, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Miodovnik, A.; Engel, S.M.; Zhu, C.; Ye, X.; Soorya, L.V.; Silva, M.J.; Calafat, A.M.; Wolff, M.S. Endocrine disruptors and childhood social impairments. Neurotoxicology 2011, 32, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.M.; Kalkbrenner, A.E.; Just, A.C.; Yolton, K.; Calafat, A.M.; Sjödin, A.; Hauser, R.; Webster, G.M.; Chen, A.; Lanphear, B.P. Gestational exposure to endocrine-disrupting chemicals and reciprocal social, repetitive, and stereotypic behaviors in 4- and 5-year-old children: The HOME study. Environ. Health Perspect. 2014, 122, 513–520. [Google Scholar] [PubMed]
- Spearow, J.L.; Doemeny, P.; Sera, R.; Leffler, R.; Barkley, M. Genetic variability in susceptibility to endocrine disruptors in mice. Science 1999, 285, 1259–1261. [Google Scholar] [CrossRef] [PubMed]
- Henningsson, S.; Jonsson, L.; Ljunggren, E.; Westberg, L.; Gillberg, C.; Råstam, M.; Anckarsäter, H.; Nygren, G.; Landén, M.; Thuresson, K.; et al. Possible association between the androgen receptor gene and autism spectrum disorder. Psychoneuroendocrinology 2009, 34, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Zettergren, A.; Jonsson, L.; Johansson, D.; Melke, J.; Lundström, S.; Anckarsäter, H.; Lichtenstein, P.; Westberg, L. Associations between polymorphisms in sex steroid related genes and autistic-like traits. Psychoneuroendocrinology 2013, 38, 2575–2584. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Delatorre, R.; Taylor, H.; Slattery, J.; Melnyk, S.; Chowdhury, N.; James, S.J. Redox metabolic abnormalities in autistic children associated with mitochondrial disease. Transl. Psychiatry 2013, 3, e273. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Boccuto, L.; Chen, C.-F.; Pittman, A.R.; Skinner, C.D.; McCartney, H.J.; Jones, K.; Bochner, B.R.; Stevenson, R.E.; Schwartz, C.E. Decreased tryptophan metabolism in patients with autism spectrum disorders. Mol. Autism 2013, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Harris, R.A.; Cheung, S.W.; Coarfa, C.; Jeong, M.; Goodell, M.A.; White, L.D.; Patel, A.; Kang, S.-H.; Shaw, C.; et al. Genomic hypomethylation in the human germline assosiates with selective structural mutability in the human genome. PLoS Genet. 2012, 8, e1002692. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Okamoto, K.-I.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Melnyk, S.; Jernigan, S.; Cleves, M.A.; Halsted, C.H.; Wong, D.H.; Cutler, P.; Bock, K.; Boris, M.; Bradstreet, J.J.; et al. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am. J. Med. Genet. Part B 2006, 141B, 947–956. [Google Scholar] [CrossRef]
- Schwartz, C.E. Aberrant tryptophan metabolism: The unifying biochemical basis for autism spectrum disorders? Biomark. Med. 2014, 8, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Frye, R.E.; Slattery, J.; Wynne, R.; Tippett, M.; Pavliv, O.; Melnyk, S.; James, S.J. Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort. PLoS ONE 2014, 9, e85436. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Frye, R.E.; Slattery, J.; Wynne, R.; Tippett, M.; Melnyk, S.; James, S.J. Oxidative stress induces mitochondrial dysfunction in a subset of autistic lymphoblastoid cell lines. Transl. Psychiatry 2014, 4, e377. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Chauhan, V.; Gu, F.; Chauhan, A. Bisphenol A induces oxidative stress and mitochondrial dysfunction in lymphoblasts from children with autism and unaffected siblings. Free Radic. Biol. Med. 2014, 76C, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Ross-Inta, C.; Wong, S.; Hung, C.; Fujisawa, Y.; Sakaguchi, D.; Angelastro, J.; Omanska-Klusek, A.; Schoenfeld, R.; Giulivi, C. Mitochondrial dysfunction in Pten haplo-insufficient mice with social deficits and repetitive behavior: Interplay between Pten and p53. PLoS ONE 2012, 7, e42504. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.W.; Mullen, G.P.; McManus, J.R.; Heatherly, J.M.; Duke, A.; Rand, J.B. Neuroligin-deficient mutants of C. elegans have sensory processing deficits and are hypersensitive to oxidative stress and mercury toxicity. Dis. Model. Mech. 2010, 3, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Pu, D.; Shen, Y.; Wu, J. Association between MTHFR gene polymorphisms and the risk of autism spectrum disorders: A meta-analysis. Autism Res. 2013, 6, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Kovač, J.; Macedoni Lukšič, M.; Trebušak Podkrajšek, K.; Klančar, G.; Battelino, T. Rare single nucleotide polymorphisms in the regulatory regions of the superoxide dismutase genes in autism spectrum disorder. Autism Res. 2014, 7, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Flodman, P.L.; Gargus, J.J.; Simon, M.T.; Verrell, K.; Haas, R.; Reiner, G.E.; Naviaux, R.; Osann, K.; Spence, M.A.; et al. Mitochondrial and Ion Channel gene alterations in autism. Biochim. Biophys. Acta 2012, 1817, 1796–1802. [Google Scholar] [CrossRef] [PubMed]
- Nava, C.; Lamari, F.; Héron, D.; Mignot, C.; Rastetter, A.; Keren, B.; Cohen, D.; Faudet, A.; Bouteiller, D.; Gilleron, M.; et al. Analysis of the chromosome X exome in patients with autism spectrum disorders identified novel candidate genes, including TMLHE. Transl. Psychiatry 2012, 2, e179. [Google Scholar] [CrossRef] [PubMed]
- Napolioni, V.; Persico, A.M.; Porcelli, V.; Palmieri, L. The mitochondrial aspartate/glutamate carrier AGC1 and calcium homeostasis: Physiological links and abnormalities in autism. Mol. Neurobiol. 2011, 44, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. A review of research trends in physiological abnormalities in autism spectrum disorders: Immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol. Psychiatry 2012, 17, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, X.; Li, X.-L.; Towers, A.; Cao, X.; Wang, P.; Bowman, R.; Yang, H.; Goldstein, J.; Li, Y.-J.; et al. Epigenetic dysregulation of SHANK3 in brain tissues from individuals with autism spectrum disorders. Hum. Mol. Genet. 2014, 23, 1563–1578. [Google Scholar] [CrossRef] [PubMed]
- Muotri, A.R.; Chu, V.T.; Marchetto, M.C.N.; Deng, W.; Moran, J.V.; Gage, F.H. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 2005, 435, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Terasaki, N.; Goodier, J.L.; Cheung, L.E.; Wang, Y.J.; Kajikawa, M.; Kazazian, H.H.; Okada, N. In vitro screening for compounds that enhance human L1 mobilization. PLoS ONE 2013, 8, e74629. [Google Scholar] [CrossRef] [PubMed]
- Muotri, A.R.; Marchetto, M.C.N.; Coufal, N.G.; Oefner, R.; Yeo, G.; Nakashima, K.; Gage, F.H. L1 retrotransposition in neurons is modulated by MeCP2. Nature 2010, 468, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Okudaira, N.; Okamura, T.; Tamura, M.; Iijma, K.; Goto, M.; Matsunaga, A.; Ochiai, M.; Nakagama, H.; Kano, S.; Fujii-Kuriyama, Y.; et al. Long interspersed element-1 is differentially regulated by food-borne carcinogens via the aryl hydrocarbon receptor. Oncogene 2013, 32, 4903–4912. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm-Benartzi, C.S.; Houseman, E.A.; Maccani, M.A.; Poage, G.M.; Koestler, D.C.; Langevin, S.M.; Gagne, L.A.; Banister, C.E.; Padbury, J.F.; Marsit, C.J. In utero exposures, infant growth, and DNA methylation of repetitive elements and developmentally related genes in human placenta. Environ. Health Perspect. 2012, 120, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Mefford, H.C.; Eichler, E.E. Duplication hotspots, rare genomic disorders, and common disease. Curr. Opin. Genet. Dev. 2009, 19, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Girirajan, S.; Johnson, R.L.; Tassone, F.; Balciuniene, J.; Katiyar, N.; Fox, K.; Baker, C.; Srikanth, A.; Yeoh, K.H.; Khoo, S.J.; et al. Global increases in both common and rare copy number load associated with autism. Hum. Mol. Genet. 2013, 22, 2870–2880. [Google Scholar] [CrossRef] [PubMed]
- Ionita-Laza, I.; Xu, B.; Makarov, V.; Buxbaum, J.D.; Roos, J.L.; Gogos, J.A.; Karayiorgou, M. Scan statistic-based analysis of exome sequencing data identifies FAN1 at 15q13.3 as a susceptibility gene for schizophrenia and autism. Proc. Natl. Acad. Sci. USA 2014, 111, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Goines, P.; van de Water, J. The immune system’s role in the biology of autism. Curr. Opin. Neurol. 2010, 23, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Onore, C.; Careaga, M.; Ashwood, P. The role of immune dysfunction in the pathophysiology of autism. Brain. Behav. Immun. 2012, 26, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Deverman, B.E.; Patterson, P.H. Cytokines and CNS development. Neuron 2009, 64, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Basil, P.; Li, Q.; Dempster, E.L.; Mill, J.; Sham, P.C.; Wong, C.C.; McAlonan, G.M. Prenatal maternal immune activation causes epigenetic differences in adolescent mouse brain. Transl. Psychiatry 2014, 2, e434. [Google Scholar] [CrossRef]
- Ashwood, P.; Schauer, J.; Pessah, I.N.; van de Water, J. Preliminary evidence of the in vitro effects of BDE-47 on innate immune responses in children with autism spectrum disorders. J. Neuroimmunol. 2009, 208, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Schwartzer, J.J.; Careaga, M.; Onore, C.E.; Rushakoff, J.A.; Berman, R.F.; Ashwood, P. Maternal immune activation and strain specific interactions in the development of autism-like behaviors in mice. Transl. Psychiatry 2013, 3, e240. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, D.; Sano, Y.; de Vries, P.J.; Dies, K.; Franz, D.; Geschwind, D.H.; Kaur, M.; Lee, Y.-S.; Li, W.; Lowe, J.K.; et al. Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Mol. Psychiatry 2012, 17, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Mercante, J.W.; Neish, A.S. Reactive oxygen production induced by the gut microbiota: Pharmacotherapeutic implications. Curr. Med. Chem. 2012, 19, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Molitoris, D.; Song, Y.; Liu, C.; Vaisanen, M.-L.; Bolte, E.; McTeague, M.; Sandler, R.; Wexler, H.; Marlowe, E.M.; et al. Gastrointestinal microflora studies in late-onset autism. Clin. Infect. Dis. 2002, 35, S6–S16. [Google Scholar] [CrossRef] [PubMed]
- Parracho, H.M.R.T.; Bingham, M.O.; Gibson, G.R.; McCartney, A.L. Differences between the gut microflora of children with autism spectrum disorders and that of healthy children. J. Med. Microbiol. 2005, 54, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.; Li, J.V.; Marchesi, J.R.; Nicholson, J.K. Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab. 2012, 16, 559–564. [Google Scholar] [CrossRef] [PubMed]
- McKnite, A.M.; Perez-Munoz, M.E.; Lu, L.; Williams, E.G.; Brewer, S.; Andreux, P.A.; Bastiaansen, J.W.M.; Wang, X.; Kachman, S.D.; Auwerx, J.; et al. Murine gut microbiota is defined by host genetics and modulates variation of metabolic traits. PLoS ONE 2012, 7, e39191. [Google Scholar] [CrossRef] [PubMed]
- Parks, B.W.; Nam, E.; Org, E.; Kostem, E.; Norheim, F.; Hui, S.T.; Pan, C.; Civelek, M.; Rau, C.D.; Bennett, B.J.; et al. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab. 2013, 17, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, J.T.; Edwards, M.; Shetty, S.R.J.; Gatewood, J.D.; Taylor, J.A.; Rissman, E.F.; Connelly, J.J. Gestational exposure to bisphenol a produces transgenerational changes in behaviors and gene expression. Endocrinology 2012, 153, 3828–3838. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.P.; Brunet, A. Bridging the transgenerational gap with epigenetic memory. Trends Genet. 2013, 29, 176–186. [Google Scholar] [CrossRef] [PubMed]
- De Theije, C.G.M.; Wopereis, H.; Ramadan, M.; van Eijndthoven, T.; Lambert, J.; Knol, J.; Garssen, J.; Kraneveld, A.D.; Oozeer, R. Altered gut microbiota and activity in a murine model of autism spectrum disorders. Brain. Behav. Immun. 2014, 37, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Viscidi, E.W.; Triche, E.W.; Pescosolido, M.F.; McLean, R.L.; Joseph, R.M.; Spence, S.J.; Morrow, E.M. Clinical characteristics of children with epilepsy and co-occuring autism. PLoS ONE 2013, 8, e67797. [Google Scholar] [CrossRef] [PubMed]
- Van Eeghen, A.M.; Pulsifer, M.B.; Merker, V.L.; Neumeyer, A.M.; van Eeghen, E.E.; Thibert, R.L.; Cole, A.J.; Leigh, F.A.; Plotkin, S.R.; Thiele, E.A. Understanding relationships between autism, intelligence, and epilepsy: A cross-disorder approach. Dev. Med. Child Neurol. 2013, 55, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Hara, H. Autism and epilepsy: A retrospective follow-up study. Brain Dev. 2007, 29, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.; Hoem, G.; Hagerman, P. Fragile X and autism: Intertwined at the molecular level leading to targeted treatments. Mol. Autism 2010, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Folsom, T.D. Dysregulation of fragile × mental retardation protein and metabotropic glutamate receptor 5 in superior frontal cortex of individuals with autism: A postmortem brain study. Mol Autism. 2011, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rustan, O.G.; Folsom, T.D.; Yousefi, M.K.; Fatemi, S.H. Phosphorylated fragile X mental retardation protein at serine 499, is reduced in cerebellar vermis and superior frontal cortex of subjects with autism: Implications for fragile X mental retardation protein-metabotropic glutamate receptor 5 signaling. Mol. Autism. 2013, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Bernard, P.B.; Castano, A.M.; O’Leary, H.; Simpson, K.; Browning, M.D.; Benke, T.A. Phosphorylation of FMRP and alterations of FMRP complex underlie enhanced mLTD in adult rats triggered by early life seizures. Neurobiol. Dis. 2013, 59, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, P.; Landrigan, P.J. Developmental neurotoxicity of industrial chemicals. Lancet 2006, 368, 2167–2178. [Google Scholar] [CrossRef] [PubMed]
- Miodovnik, A. Environmental neurotoxicants and the developing brain. Mt. Sinai J. Med. 2011, 78, 58–77. [Google Scholar] [CrossRef] [PubMed]
- Volk, H.E.; Kerin, T.; Lurmann, F.; Hertz-Picciotto, I.; McConnell, R.; Campbell, D.B. Autism spectrum disorder: Interaction of air pollution with the MET receptor tyrosine kinase gene. Epidemiology 2014, 25, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Kohane, I.S.; Valtchinov, V.I. Quantifying the white blood cell transcriptome as an accessible window to the multiorgan transcriptome. Bioinform. Oxf. Engl. 2012, 28, 538–545. [Google Scholar] [CrossRef]
- Main, P.A.E.; Thomas, P.; Esterman, A.; Fenech, M.F. Necrosis is increased in lymphoblastoid cell lines from children with autism compared with their non-autistic siblings under conditions of oxidative and nitrosative stress. Mutagenesis 2013, 28, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Reuter, J.; Gerich, F.J.; Hildebrandt, B.; Hägele, S.; Katschinski, D.; Müller, M. Enhanced hypoxia susceptibility in hippocampal slices from a mouse model of rett syndrome. J. Neurophysiol. 2009, 101, 1016–1032. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koufaris, C.; Sismani, C. Modulation of the Genome and Epigenome of Individuals Susceptible to Autism by Environmental Risk Factors. Int. J. Mol. Sci. 2015, 16, 8699-8718. https://doi.org/10.3390/ijms16048699
Koufaris C, Sismani C. Modulation of the Genome and Epigenome of Individuals Susceptible to Autism by Environmental Risk Factors. International Journal of Molecular Sciences. 2015; 16(4):8699-8718. https://doi.org/10.3390/ijms16048699
Chicago/Turabian StyleKoufaris, Costas, and Carolina Sismani. 2015. "Modulation of the Genome and Epigenome of Individuals Susceptible to Autism by Environmental Risk Factors" International Journal of Molecular Sciences 16, no. 4: 8699-8718. https://doi.org/10.3390/ijms16048699
APA StyleKoufaris, C., & Sismani, C. (2015). Modulation of the Genome and Epigenome of Individuals Susceptible to Autism by Environmental Risk Factors. International Journal of Molecular Sciences, 16(4), 8699-8718. https://doi.org/10.3390/ijms16048699