
Phenotypic and Molecular Convergence of 2q23.1 Deletion Syndrome with Other Neurodevelopmental Syndromes Associated with Autism Spectrum Disorder


Abstract
:
1. Introduction
2. Results and Discussion
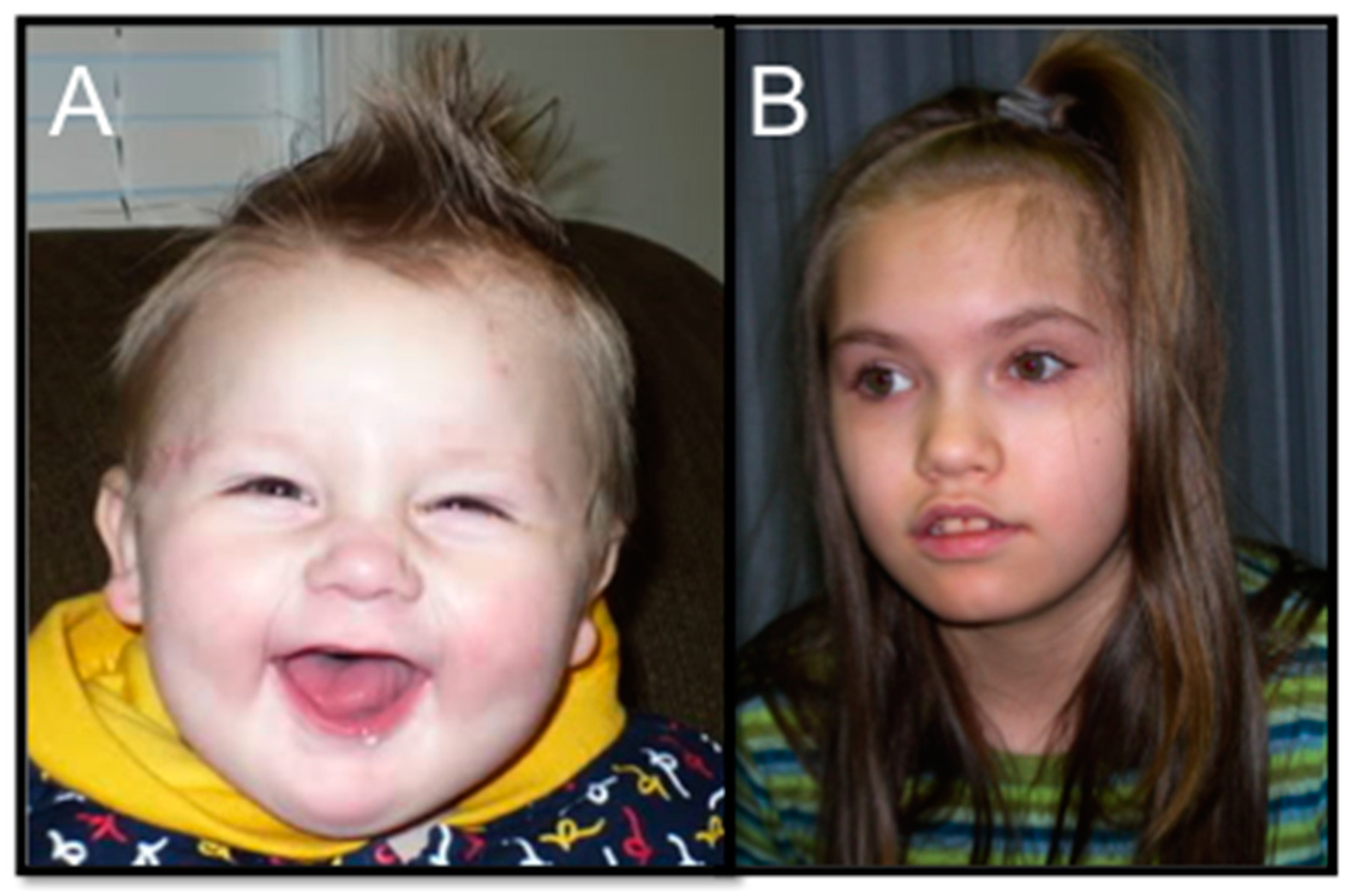
2.1. 2q23.1 Deletion Syndrome Clinical Review


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2q23.1 Deletion 1 | ||
|---|---|---|
| Common Features | Frequency | Percentage (%) |
| Neurological | ||
| Developmental delay | 74/74 | 100 |
| Motor delay | 45/45 | 100 |
| Language impairment | 51/54 | 94.4 |
| Ataxia | 22/32 | 68.7 |
| Infantile hypotonia | 28/30 | 93.3 |
| Infantile feeding difficulties | 17/20 | 85.0 |
| Seizures | 45/53 | 84.9 |
| Behavioral | ||
| Autistic-like behaviors | 60/61 | 98.4 |
| Behavioral problems | 60/61 | 98.4 |
| Aggression/temper tantrums | 13/21 | 62.9 |
| Distractibility/short attention span | 21/21 | 100 |
| Hyperphagia | 8/16 | 50.0 |
| Self-injurious behaviors | 21/33 | 63.6 |
| Sleep disturbances | 41/52 | 78.8 |
| Growth/Endocrine Abnormalities | ||
| Postnatal growth retardation | 25/51 | 49.0 |
| Obesity | 6/17 | 35.3 |
| Short stature (<5th percentile) | 30/43 | 69.8 |
| Craniofacial Abnormalities | ||
| Cranium | ||
| Brachycephaly | 12/36 | 33.3 |
| Broad forehead | 21/30 | 70.0 |
| Microcephaly | 28/46 | 60.9 |
| Eyes | ||
| Arched/thick eyebrows | 19/24 | 79.2 |
| Myopia/hypermetropia/corrective lenses | 8/11 | 72.7 |
| Synophrys | 13/28 | 46.4 |
| Nose/Ear | ||
| Nasal abnormalities | 42/43 | 97.7 |
| Outer ear abnormalities | 22/29 | 75.9 |
| Mouth/Chin | ||
| Dental abnormalities | 18/35 | 51.4 |
| Downturned corners of the mouth | 20/28 | 71.4 |
| Macroglossia or protruding tongue | 8/33 | 24.2 |
| Micrognathia/retrognathia | 16/30 | 53.3 |
| Open mouth | 26/34 | 76.5 |
| Tented upper lip | 19/30 | 63.3 |
| Thin upper lip | 21/28 | 75.0 |
| Thick or everted lower lip | 19/26 | 73.1 |
| Wide mouth | 15/25 | 60.0 |
| Skeletal Extremity Abnormalities | ||
| Brachydactyly | 14/33 | 42.4 |
| Clinodactyly, 5th finger | 24/38 | 63.2 |
| Sandal gap | 12/32 | 37.5 |
| Short fifth digit | 16/37 | 43.2 |
| Small hands and feet | 25/37 | 67.6 |
2.2. Overlapping Phenotypes across 2q23.1 Deletion Syndrome and Other Autism Spectrum Disorders
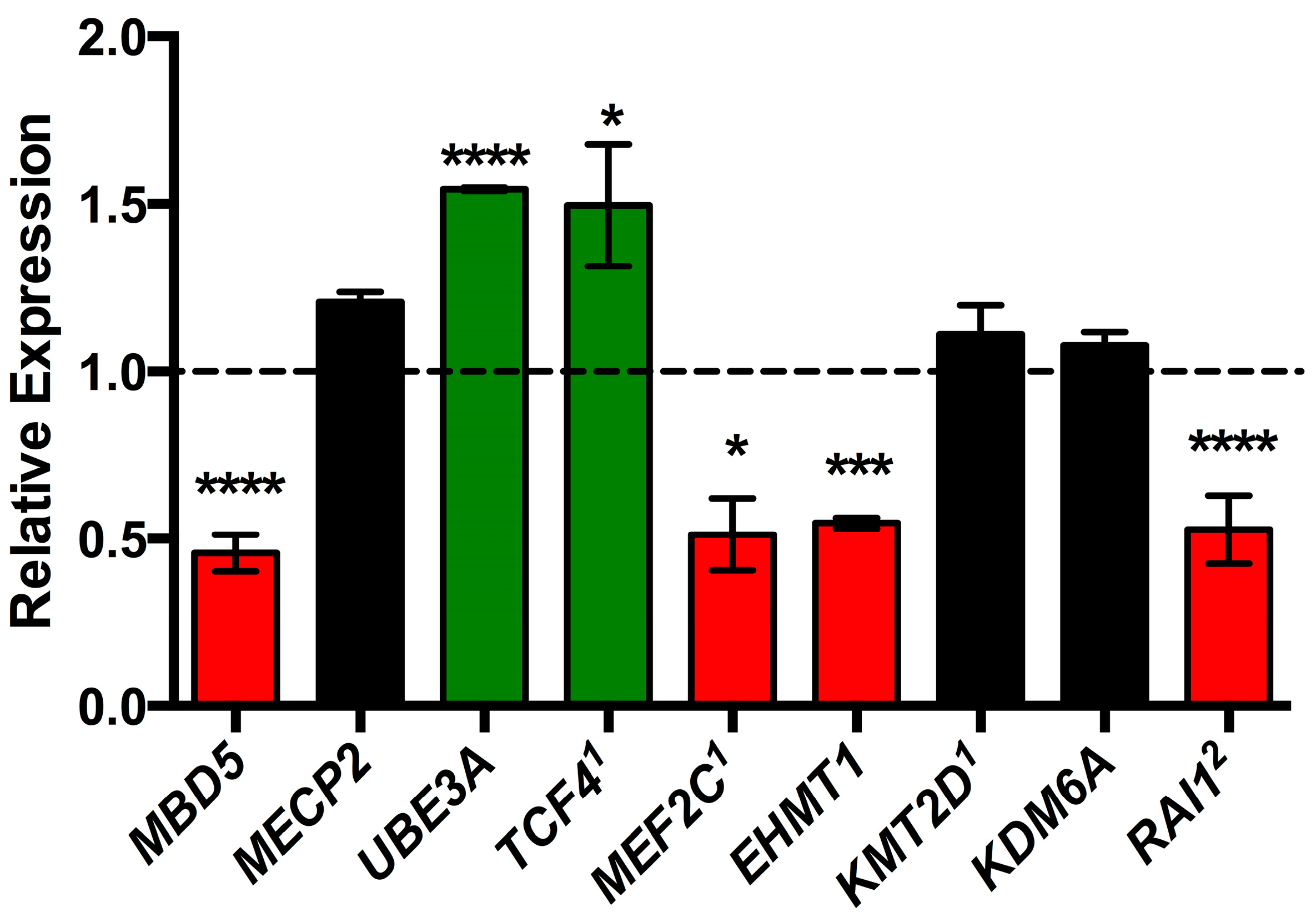
2.3. MBD5 Regulates Disorder-Specific Genes

| DISORDER | 2q23.1 del | RTT | AS | PTHS | 2q23.1 dup | 5q14.3 del | KFS | KMS | SMS |
|---|---|---|---|---|---|---|---|---|---|
| Key References | [13,20] | [33,34] | [31,35] | [36,37] | [38,39] | [40] | [41] | [42,43] | [44] |
| Causative Gene | MBD5 | MECP2 | UBE3A | TCF4 | MBD5 | MEF2C | EHMT1 | KMT2D, KDM6A | RAI1 |
| Neurological/Behavioral Characteristics | |||||||||
| Intellectual disability a | +++ | +++ | +++ | +++ | ++ | +++ | +++ | ++ | ++ |
| Speech delay b | +++ | +++ | +++ | +++ | ++ | +++ | ++ | ++ | + |
| Seizures c | +++ | +++ | +++ | ++ | ++ | ++ | ++ | + | + |
| Sleep disturbance d | +++ | + | +++ | ++ | ++ | + | + | + | +++ |
| Delayed walking e | ++ | +++ | ++ | ++ | + | +++ | + | + | + |
| Hypotonia | + | + | + | + | + | + | + | + | + |
| Autism-like behaviors | + | + | + | + | + | + | +f | + | + |
| Feeding difficulties | + | + | + | - | + | + | + | + | + |
| Stereotypic behaviors | + | + | + | + | + | + | - | + | + |
| Ataxia | + | + | + | + | + | + | - | + | - |
| Happy disposition (frequent or inappropriate laughing) | + | + | + | + | + | NR | - | + | - |
| Hyperactivity/short attention span | + | - | + | + | + | - | - | + | + |
| Self-injurious behavior | + | - | - | - | - | - | + | - | + |
| Aggressive behavior | - | - | - | + | - | - | + | - | + |
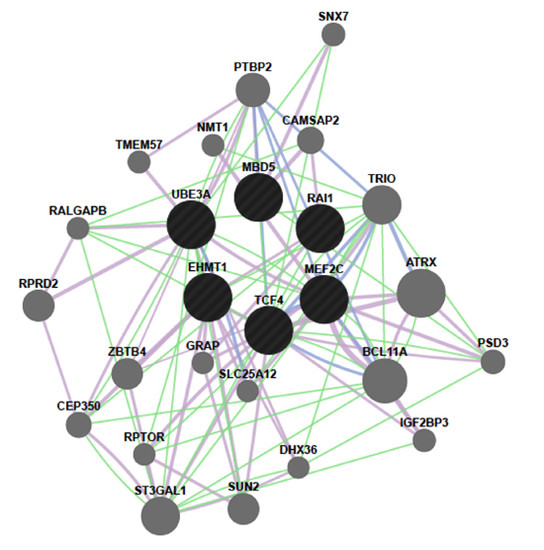
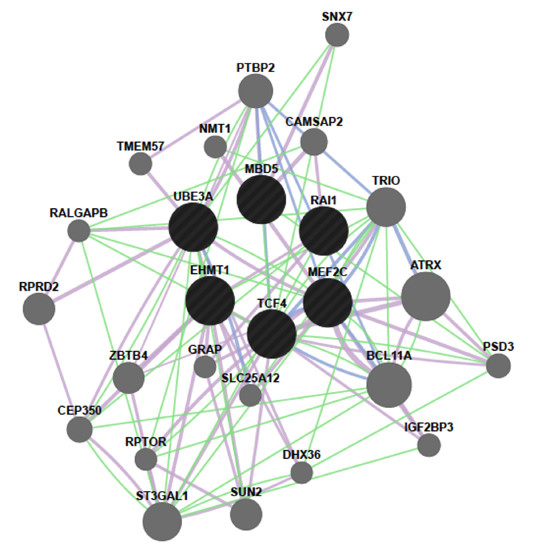
2.4. MBD5 Network
2.5. Molecular Relationships between MBD5 and RAI1

| Ingenuity Canonical Pathways * |
|---|
| Acetate conversion to acetyl-CoA |
| Agrin interactions at neuromuscular junction |
| Aldosterone signaling in epithelial cells |
| Amyotrophic lateral sclerosis signaling |
| Axonal guidance signaling |
| Cyclin-dependent kinsase 5 signaling |
| Choline degradation I |
| Circadian rhythm signaling |
| Ciliary neurotrophic factor signaling |
| Docosahexaenoic acid signaling |
| Dolichol and dolichyl phosphate biosynthesis |
| Eukaryotic initiation factor 2 signaling |
| Ephrin A signaling |
| Ephrin B signaling |
| Ephrin receptor signaling |
| Epidermal growth factor receptor signaling |
| G protein signaling mediated by Tubby |
| Gap junction signaling |
| Glial cell line-derived neurotrophic factors family ligand-receptor interactions |
| Glioma signaling |
| Melanoma signaling |
| Mechanistic target of rapamycin signaling |
| Netrin signaling |
| Neuregulin signaling |
| Ingenuity Canonical Pathways * |
|---|
| 14-3-3-mediated signaling |
| 1D-myo-inositol hexakisphosphate biosynthesis II (mammalian) |
| Acetate conversion to acetyl-CoA |
| Agrin interactions at neuromuscular junction |
| Aldosterone signaling in epithelial cells |
| Angiopoietin signaling |
| Apoptosis signaling |
| Aryl hydrocarbon receptor signaling |
| Assembly of RNA polymerase II complex |
| Assembly of RNA polymerase III complex |
| Ataxia telangiectasia mutated signaling |
| Bone morphogenetic protein signaling |
| CD40 signaling |
| Cell division control protein 42 homolog signaling |
| Cytidine diphosphate diacylglycerol-diacylglycerol biosynthesis I |
| Cell cycle control of chromosomal replication |
| Cell Cycle: G1/S checkpoint regulation |
| Cell Cycle: G2/M DNA damage checkpoint regulation |
| Ceramide signaling |
| Cholecystokinin/gastrin-mediated signaling |
| Choline degradation I |
| Circadian rhythm signaling |
| Citrulline-nitric oxide cycle |
| Clathrin-mediated endocytosis signaling |
3. Experimental Section
3.1. Patients and Cell Culture Studies
3.2. Expression Analyses
3.3. Network Analysis
3.4. Ingenuity Pathway Analysis (IPA)
3.5. Statistical Analyses
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Spooren, W.; Lindemann, L.; Ghosh, A.; Santarelli, L. Synapse dysfunction in autism: A molecular medicine approach to drug discovery in neurodevelopmental disorders. Trends Pharmacol. Sci. 2012, 33, 669–684. [Google Scholar] [CrossRef] [PubMed]
- Ceroni, F.; Sagar, A.; Simpson, N.H.; Gawthrope, A.J.; Newbury, D.F.; Pinto, D.; Francis, S.M.; Tessman, D.C.; Cook, E.H.; Monaco, A.P.; et al. A deletion involving CD38 and BST1 results in a fusion transcript in a patient with autism and asthma. Autism Res. 2014, 7, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Pendergrass, S.; Girirajan, S.; Selleck, S. Uncovering the etiology of autism spectrum disorders: Genomics, bioinformatics, environment, data collection and exploration, and future possibilities. Pac. Symp. Biocomput. 2014, 422–426. [Google Scholar]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Betancur, C. Etiological heterogeneity in autism spectrum disorders: More than 100 genetic and genomic disorders and still counting. Brain Res. 2011, 1380, 42–77. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, I.; Ragavendran, A.; Erdin, S.; Klei, L.; Sugathan, A.; Guide, J.R.; Manavalan, P.; Zhou, J.Q.; Wheeler, V.C.; Levin, J.Z.; et al. Transcriptional consequences of 16p11.2 deletion and duplication in mouse cortex and multiplex autism families. Am. J. Hum. Genet. 2014, 94, 870–883. [Google Scholar] [CrossRef] [PubMed]
- Pasciuto, E.; Bagni, C. Snapshot: Fmrp interacting proteins. Cell 2014, 159, 218–218.e1. [Google Scholar] [CrossRef] [PubMed]
- Van Bokhoven, H. Genetic and epigenetic networks in intellectual disabilities. Annu. Rev. Genet. 2011, 45, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Urdinguio, R.G.; Sanchez-Mut, J.V.; Esteller, M. Epigenetic mechanisms in neurological diseases: Genes, syndromes, and therapies. Lancet Neurol. 2009, 8, 1056–1072. [Google Scholar] [CrossRef] [PubMed]
- Oti, M.; Huynen, M.A.; Brunner, H.G. Phenome connections. Trends Genet. 2008, 24, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Vissers, L.E.; de Vries, B.B.; Osoegawa, K.; Janssen, I.M.; Feuth, T.; Choy, C.O.; Straatman, H.; van der Vliet, W.; Huys, E.H.; van Rijk, A.; et al. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am. J. Hum. Genet. 2003, 73, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Talkowski, M.E.; Mullegama, S.V.; Rosenfeld, J.A.; van Bon, B.W.; Shen, Y.; Repnikova, E.A.; Gastier-Foster, J.; Thrush, D.L.; Kathiresan, S.; Ruderfer, D.M.; et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am. J. Hum. Genet. 2011, 89, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Laget, S.; Joulie, M.; le Masson, F.; Sasai, N.; Christians, E.; Pradhan, S.; Roberts, R.J.; Defossez, P.A. The human proteins MBD5 and MBD6 associate with heterochromatin but they do not bind methylated DNA. PLoS ONE 2010, 5, e11982. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.; Ali Khan, A.; Bresso, E.; Vigouroux, C.; Beri, M.; Lejczak, S.; Deemer, B.; Andrieux, J.; Philippe, C.; Moncla, A.; et al. Extended spectrum of mbd5 mutations in neurodevelopmental disorders. Eur. J. Hum. Genet. 2013, 21, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Cukier, H.N.; Lee, J.M.; Ma, D.; Young, J.I.; Mayo, V.; Butler, B.L.; Ramsook, S.S.; Rantus, J.A.; Abrams, A.J.; Whitehead, P.L.; et al. The expanding role of mbd genes in autism: Identification of a mecp2 duplication and novel alterations in mbd5, mbd6, and setdb1. Autism Res. 2012, 5, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Liu, B.; Guo, F.; Xu, G.; Ding, Y.; Liu, Y.; Sun, X.; Xu, G. The essential role of MBD5 in the regulation of somatic growth and glucose homeostasis in mice. PLoS ONE 2012, 7, e47358. [Google Scholar] [CrossRef] [PubMed]
- Ladha, S. Getting to the bottom of autism spectrum and related disorders: MBD5 as a key contributor. Clin. Genet. 2012, 81, 363–364. [Google Scholar] [CrossRef] [PubMed]
- Mullegama, S.V.; Pugliesi, L.; Burns, B.; Shah, Z.; Tahir, R.; Gu, Y.; Nelson, D.L.; Elsea, S.H. MBD5 haploinsufficiency is associated with slee disturbance and disrupts circadian pathways common to smith-magenis and fragile X syndromes. Eur J. Hum. Genet. 2014. [Google Scholar] [CrossRef]
- Hodge, J.C.; Mitchell, E.; Pillalamarri, V.; Toler, T.L.; Bartel, F.; Kearney, H.M.; Zou, Y.S.; Tan, W.H.; Hanscom, C.; Kirmani, S.; et al. Disruption of mbd5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities. Mol. Psychiatry 2014, 19, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; An, Y.; Yu, L.; Liu, R.; Qin, Y.; Guo, X.; Sun, D.; Zhou, S.; Wu, B.; Jiang, Y.H.; et al. A genomic copy number variant analysis implicates the mbd5 and hnrnpu genes in chinese children with infantile spasms and expands the clinical spectrum of 2q23.1 deletion. BMC Med. Genet. 2014, 15, 62. [Google Scholar] [CrossRef] [PubMed]
- Lund, C.; Brodtkorb, E.; Rosby, O.; Rodningen, O.K.; Selmer, K.K. Copy number variants in adult patients with lennox-gastaut syndrome features. Epilepsy Res. 2013, 105, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Shichiji, M.; Ito, Y.; Shimojima, K.; Nakamu, H.; Oguni, H.; Osawa, M.; Yamamoto, T. A cryptic microdeletion including MBD5 occurring within the breakpoint of a reciprocal translocation between chromosomes 2 and 5 in a patient with developmental delay and obesity. Am. J. Med. Genet. A 2013, 161, 850–855. [Google Scholar] [CrossRef]
- Girirajan, S.; Dennis, M.Y.; Baker, C.; Malig, M.; Coe, B.P.; Campbell, C.D.; Mark, K.; Vu, T.H.; Alkan, C.; Cheng, Z.; et al. Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am. J. Hum. Genet. 2013, 92, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.R.; Mullegama, S.V.; Rosenfeld, J.A.; Dagli, A.I.; Hatchwell, E.; Allen, W.P.; Williams, C.A.; Elsea, S.H. Haploinsufficiency of mbd5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. Eur. J. Hum. Genet. 2010, 18, 436–441. [Google Scholar] [CrossRef]
- Van Bon, B.W.; Koolen, D.A.; Brueton, L.; McMullan, D.; Lichtenbelt, K.D.; Ades, L.C.; Peters, G.; Gibson, K.; Moloney, S.; Novara, F.; et al. The 2q23.1 microdeletion syndrome: Clinical and behavioural phenotype. Eur J. Hum. Genet. 2010, 18, 163–170. [Google Scholar]
- Noh, G.J.; Graham, J.M., Jr. 2q23.1 microdeletion of the MBD5 gene in a female with seizures, developmental delay and distinct dysmorphic features. Eur. J. Med. Genet. 2012, 55, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Motobayashi, M.; Nishimura-Tadaki, A.; Inaba, Y.; Kosho, T.; Miyatake, S.; Niimi, T.; Nishimura, T.; Wakui, K.; Fukushima, Y.; Matsumoto, N.; et al. Neurodevelopmental features in 2q23.1 microdeletion syndrome: Report of a new patient with intractable seizures and review of literature. Am. J. Med. Genet. A 2012, 158, 861–868. [Google Scholar] [CrossRef]
- Chung, B.H.; Stavropoulos, J.; Marshall, C.R.; Weksberg, R.; Scherer, S.W.; Yoon, G. 2q23 de novo microdeletion involving the mbd5 gene in a patient with developmental delay, postnatal microcephaly and distinct facial features. Am. J. Med. Genet. A 2011, 155, 424–429. [Google Scholar] [CrossRef]
- Wagenstaller, J.; Spranger, S.; Lorenz-Depiereux, B.; Kazmierczak, B.; Nathrath, M.; Wahl, D.; Heye, B.; Glaser, D.; Liebscher, V.; Meitinger, T.; et al. Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am. J. Hum. Genet. 2007, 81, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.H.; Bird, L.M.; Thibert, R.L.; Williams, C.A. If not angelman, what is it? A review of angelman-like syndromes. Am. J. Med. Genet. A 2014, 164, 975–992. [Google Scholar] [CrossRef]
- Kleefstra, T.; Kramer, J.M.; Neveling, K.; Willemsen, M.H.; Koemans, T.S.; Vissers, L.E.; Wissink-Lindhout, W.; Fenckova, M.; van den Akker, W.M.; Kasri, N.N.; et al. Disruption of an ehmt1-associated chromatin-modification module causes intellectual disability. Am. J. Hum. Genet. 2012, 91, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Smeets, E.E.; Pelc, K.; Dan, B. Rett syndrome. Mol. Syndromol. 2012, 2, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L. The relationship of Rett syndrome and MECP2 disorders to autism. Dialogues Clin. Neurosci. 2012, 14, 253–262. [Google Scholar] [PubMed]
- Kyllerman, M. Angelman syndrome. Handb. Clin. Neurol. 2013, 111, 287–290. [Google Scholar] [PubMed]
- Whalen, S.; Heron, D.; Gaillon, T.; Moldovan, O.; Rossi, M.; Devillard, F.; Giuliano, F.; Soares, G.; Mathieu-Dramard, M.; Afenjar, A.; et al. Novel comprehensive diagnostic strategy in Pitt-hopkins syndrome: Clinical score and further delineation of the TCF4 mutational spectrum. Hum. Mutat. 2012, 33, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Marangi, G.; Ricciardi, S.; Orteschi, D.; Tenconi, R.; Monica, M.D.; Scarano, G.; Battaglia, D.; Lettori, D.; Vasco, G.; Zollino, M. Proposal of a clinical score for the molecular test for Pitt-hopkins syndrome. Am. J. Med. Genet. A 2012, 158, 1604–1611. [Google Scholar] [CrossRef]
- Chung, B.H.; Mullegama, S.; Marshall, C.R.; Lionel, A.C.; Weksberg, R.; Dupuis, L.; Brick, L.; Li, C.; Scherer, S.W.; Aradhya, S.; et al. Severe intellectual disability and autistic features associated with microduplication 2q23.1. Eur. J. Hum. Genet. 2012, 20, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Mullegama, S.V.; Rosenfeld, J.A.; Orellana, C.; van Bon, B.W.; Halbach, S.; Repnikova, E.A.; Brick, L.; Li, C.; Dupuis, L.; Rosello, M.; et al. Reciprocal deletion and duplication at 2q23.1 indicates a role for MBD5 in autism spectrum disorder. Eur. J. Hum. Genet. 2014, 22, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Novara, F.; Rizzo, A.; Bedini, G.; Girgenti, V.; Esposito, S.; Pantaleoni, C.; Ciccone, R.; Sciacca, F.L.; Achille, V.; Della Mina, E.; et al. Mef2c deletions and mutations versus duplications: A clinical comparison. Eur. J. Med. Genet. 2013, 56, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, M.H.; Vulto-van Silfhout, A.T.; Nillesen, W.M.; Wissink-Lindhout, W.M.; van Bokhoven, H.; Philip, N.; Berry-Kravis, E.M.; Kini, U.; van Ravenswaaij-Arts, C.M.; Delle Chiaie, B.; et al. Update on Kleefstra syndrome. Mol. Syndromol. 2012, 2, 202–212. [Google Scholar] [PubMed]
- Banka, S.; Lederer, D.; Benoit, V.; Jenkins, E.; Howard, E.; Bunstone, S.; Kerr, B.; McKee, S.; Lloyd, I.C.; Shears, D.; et al. Novel KDM6A (UTX) mutations and a clinical and molecular review of the X-linked Kabuki syndrome (KS2). Clin. Genet. 2015, 87, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Bogershausen, N.; Wollnik, B. Unmasking Kabuki syndrome. Clin. Genet. 2013, 83, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Elsea, S.H.; Girirajan, S. Smith-magenis syndrome. Eur. J. Hum. Genet. 2008, 16, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabasi, A.L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.I.; Choi, I.G. Exploring the human diseasome: The human disease network. Brief. Funct. Genomics 2012, 11, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Jupiter, D.C.; VanBuren, V. A visual data mining tool that facilitates reconstruction of transcription regulatory networks. PLoS ONE 2008, 3, e1717. [Google Scholar] [CrossRef] [PubMed]
- Duhr, F.; Deleris, P.; Raynaud, F.; Seveno, M.; Morisset-Lopez, S.; Mannoury la Cour, C.; Millan, M.J.; Bockaert, J.; Marin, P.; Chaumont-Dubel, S. Cdk5 induces constitutive activation of 5-HT6 receptors to promote neurite growth. Nat. Chem. Biol. 2014, 10, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Camarena, V.; Cao, L.; Abad, C.; Abrams, A.; Toledo, Y.; Araki, K.; Araki, M.; Walz, K.; Young, J.I. Disruption of mbd5 in mice causes neuronal functional deficits and neurobehavioral abnormalities consistent with 2q23.1 microdeletion syndrome. EMBO Mol. Med. 2014, 6, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Tahir, R.; Kennedy, A.; Elsea, S.H.; Dickinson, A.J. Retinoic acid induced-1 (Rai1) regulates craniofacial and brain development in Xenopus. Mech. Dev. 2014, 133, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Alberts, I.; Li, X. The apoptotic perspective of autism. Int. J. Dev. Neurosci. 2014, 36, 13–18. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mullegama, S.V.; Alaimo, J.T.; Chen, L.; Elsea, S.H. Phenotypic and Molecular Convergence of 2q23.1 Deletion Syndrome with Other Neurodevelopmental Syndromes Associated with Autism Spectrum Disorder. Int. J. Mol. Sci. 2015, 16, 7627-7643. https://doi.org/10.3390/ijms16047627
Mullegama SV, Alaimo JT, Chen L, Elsea SH. Phenotypic and Molecular Convergence of 2q23.1 Deletion Syndrome with Other Neurodevelopmental Syndromes Associated with Autism Spectrum Disorder. International Journal of Molecular Sciences. 2015; 16(4):7627-7643. https://doi.org/10.3390/ijms16047627
Chicago/Turabian StyleMullegama, Sureni V., Joseph T. Alaimo, Li Chen, and Sarah H. Elsea. 2015. "Phenotypic and Molecular Convergence of 2q23.1 Deletion Syndrome with Other Neurodevelopmental Syndromes Associated with Autism Spectrum Disorder" International Journal of Molecular Sciences 16, no. 4: 7627-7643. https://doi.org/10.3390/ijms16047627
APA StyleMullegama, S. V., Alaimo, J. T., Chen, L., & Elsea, S. H. (2015). Phenotypic and Molecular Convergence of 2q23.1 Deletion Syndrome with Other Neurodevelopmental Syndromes Associated with Autism Spectrum Disorder. International Journal of Molecular Sciences, 16(4), 7627-7643. https://doi.org/10.3390/ijms16047627