
Humic Acid Increases Amyloid β-Induced Cytotoxicity by Induction of ER Stress in Human SK-N-MC Neuronal Cells


Abstract
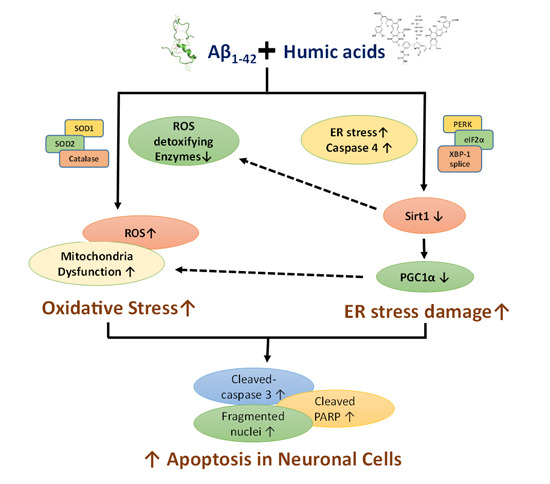
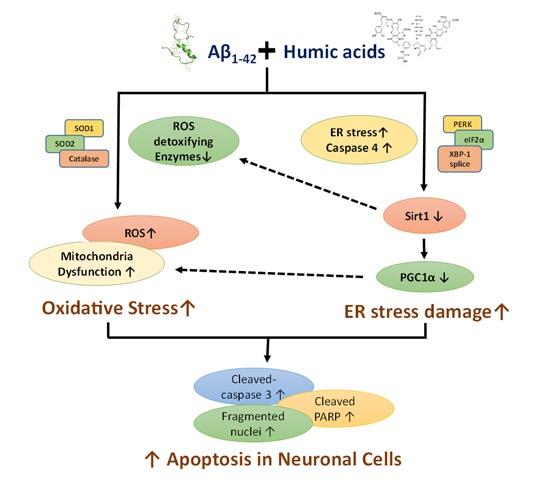
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
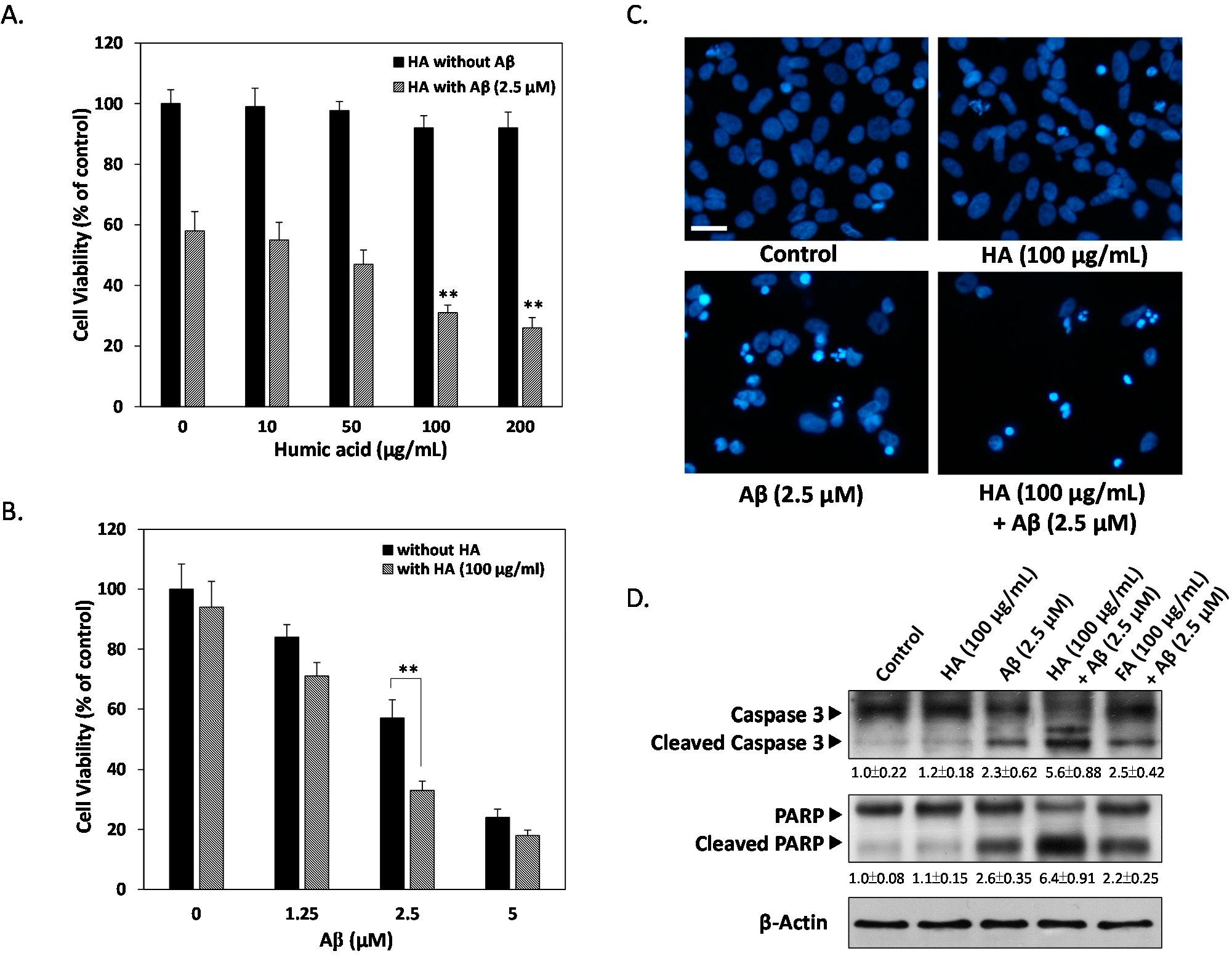
2.1. Effects of Humic Acid (HA) on Aβ-Induced Cytotoxicity



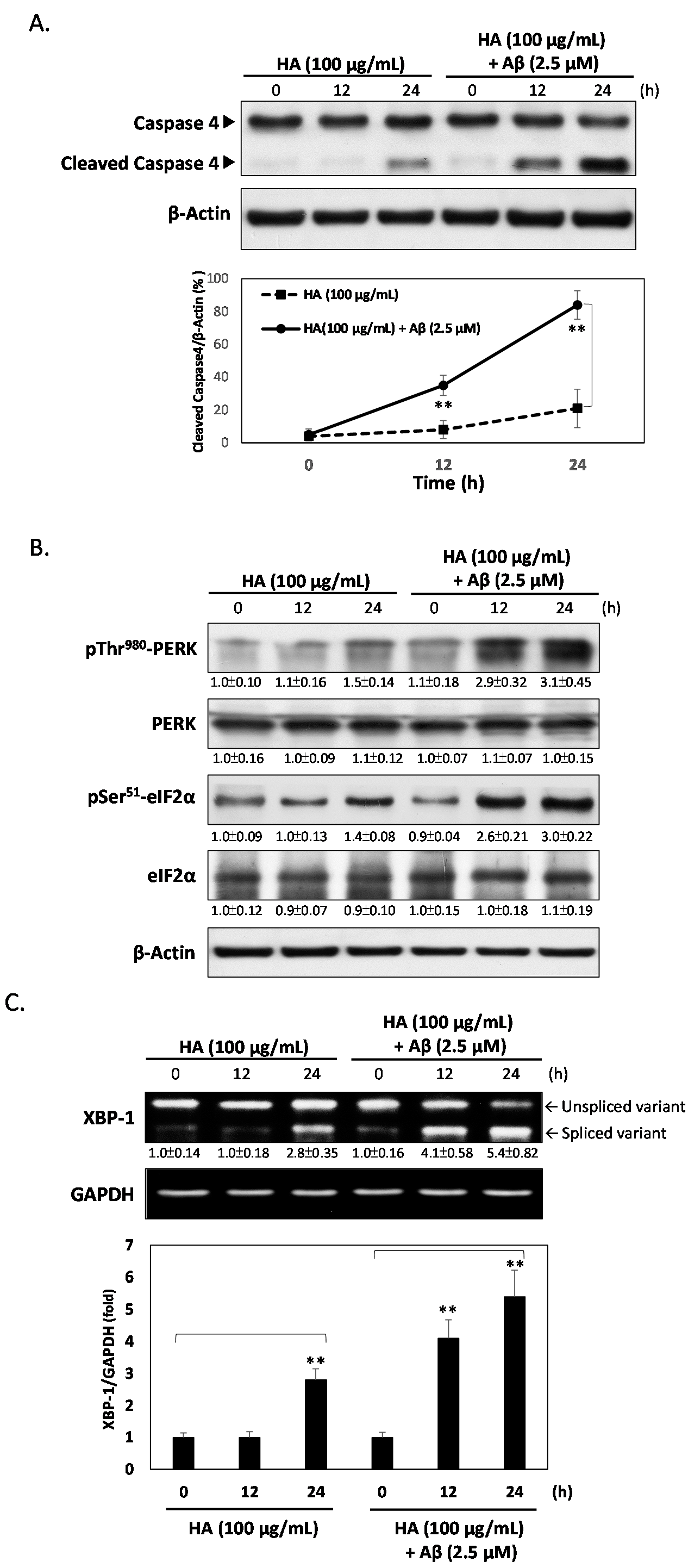
2.2. HA Increases Aβ-Induced Endoplasmic Reticulum (ER) Stress
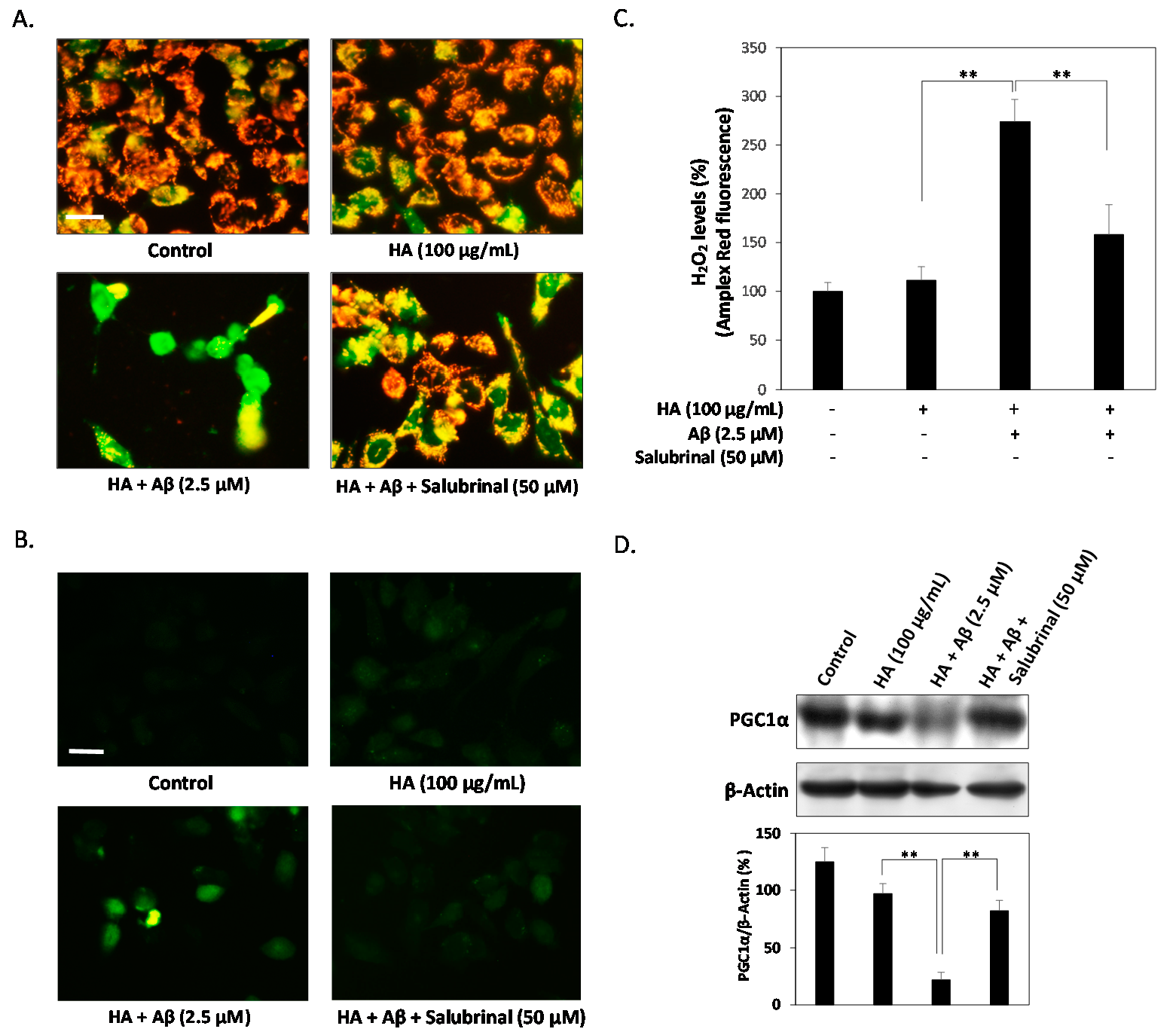
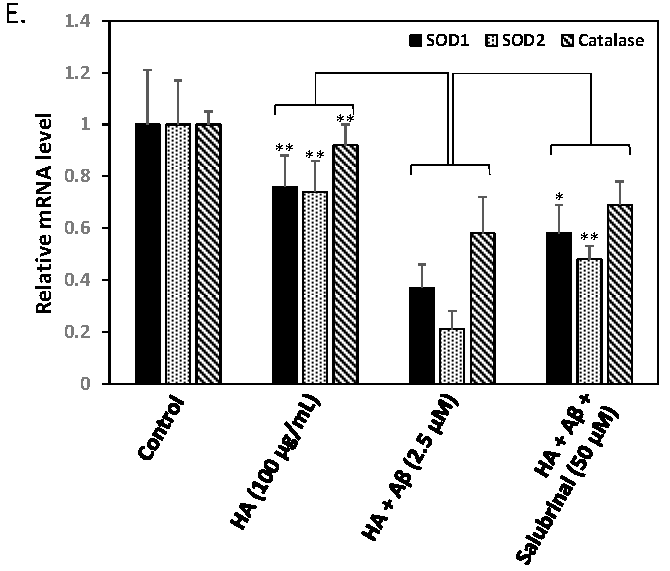
2.3. HA Exacerbates Aβ-Induced Mitochondria Dysfunction and ROS Accumulation


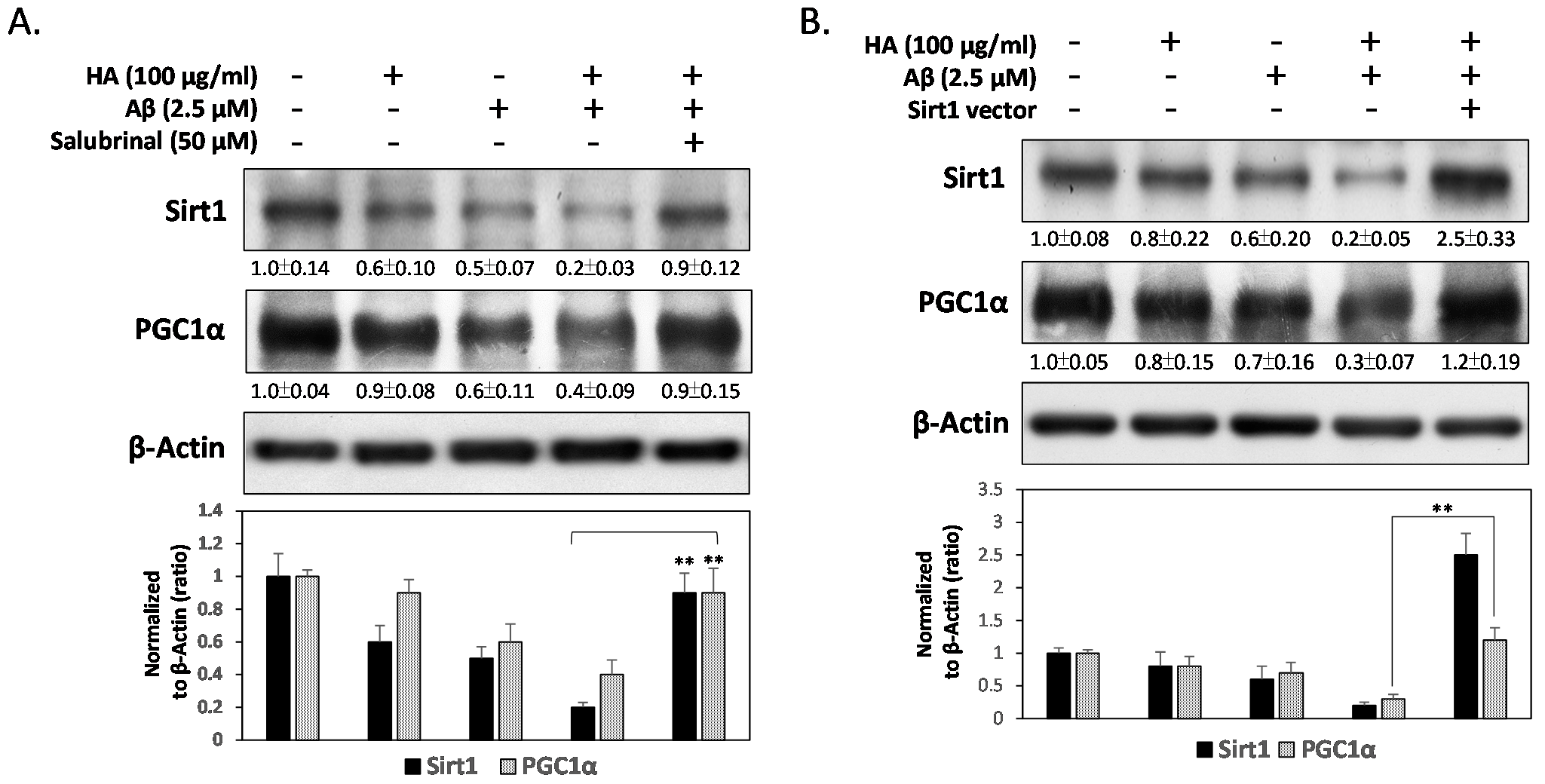
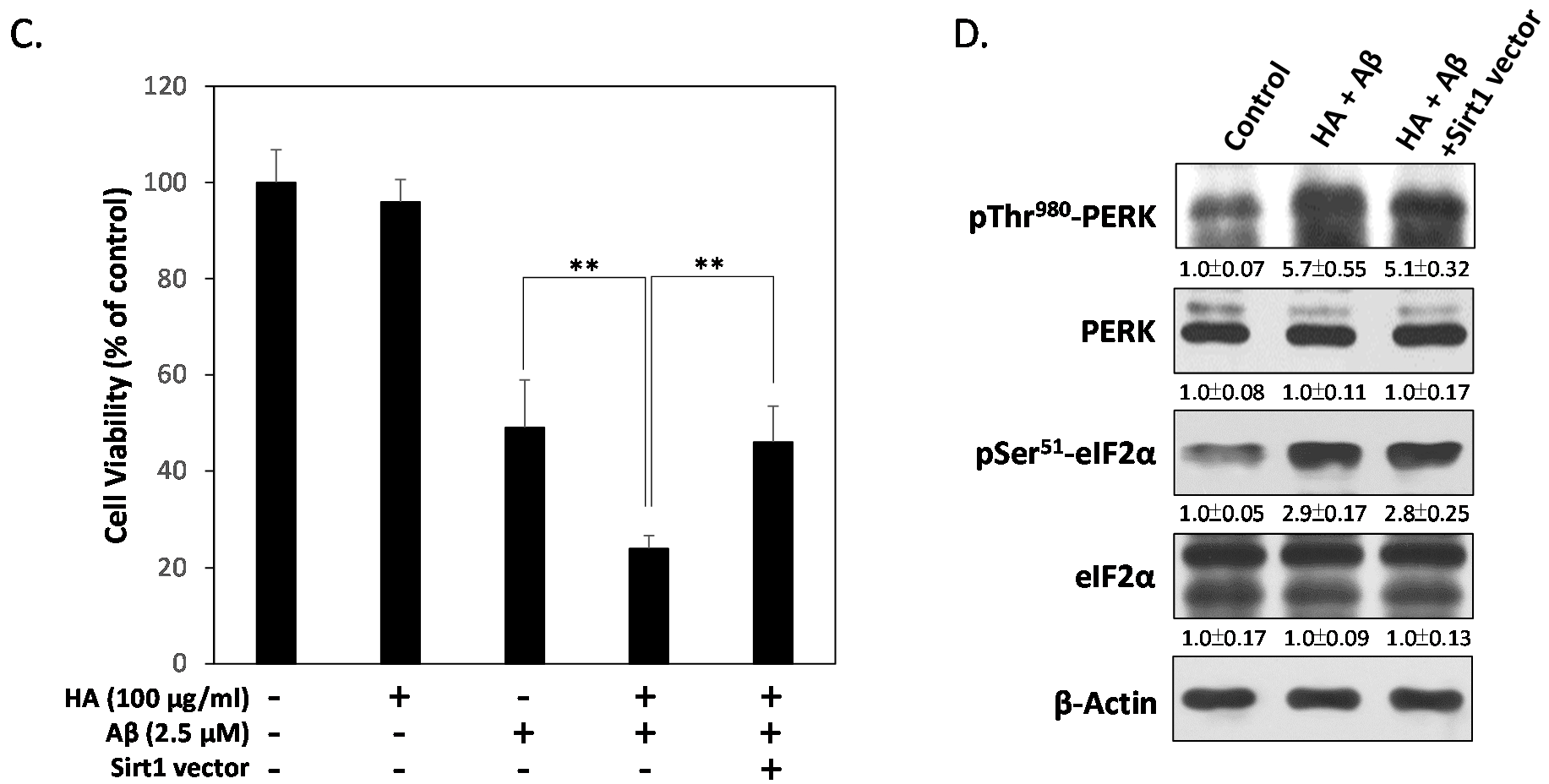
2.4. HA and Aβ-Induced ER Stress Disturbs Sirt1/PGC1α Signaling


3. Discussion
4. Experimental Section
4.1. Materials
4.2. Preparation of Synthetic Humic Acid (HA)
4.3. Cell Culture and Viability Assay
4.4. Nuclei Morphology by 4',6-Diamidino-2-phenylindole (DAPI) Staining
4.5. Western Blot Analysis
4.6. Reverse Transcription Polymerase Chain Reaction (RT-PCR) Analysis of XBP1 mRNA Splicing
4.7. Analysis of Mitochondrial Membrane Potential
4.8. Measurement of Reactive Oxygen Species (ROS)
4.9. Real Time Quantitative PCR Analysis of mRNA Expression
4.10. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schulten, H.R.; Schnitzer, M. A state of the art structural concept for humic substances. Naturwissenschaften 1993, 80, 29–30. [Google Scholar] [CrossRef]
- Schmidt, G.; Pesch, R.; Schröder, W.; Conrad, A.; Kolossa-Gehring, M.; Feigenspan, S.; Dobler, L.; Wiesmüller, G.A.; Birke, M.; Utermann, J. The potential of spatial information in human biomonitoring by example of two German environmental epidemiology studies. Environ. Geochem. Health 2011, 33, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.J. Blackfoot disease: Arsenic or humic acid? Lancet 1990, 336, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Gau, R.J.; Yang, H.L.; Suen, J.L.; Lu, F.J. Induction of oxidative stress by humic acid through increasing intracellular iron: A possible mechanism leading to atherothrombotic vascular disorder in blackfoot disease. Biochem. Biophys. Res. Commun. 2001, 283, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Briasoulis, A.; Papageorgiou, N.; Tsioufis, C.; Tsiamis, E.; Toutouzas, K.; Stefanadis, C. Oxidative stress and endothelial function: Therapeutic interventions. Recent Pat. Cardiovasc. Drug Discov. 2011, 6, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Das, J.R.; Eberhardt, R.T. Contemporary risk assessment and cardiovascular outcomes in peripheral arterial disease. Cardiovasc. Hematol. Disord. Drug Targets 2013, 13, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Rafnsson, S.B.; Deary, I.J.; Fowkes, F.G. Peripheral arterial disease and cognitive function. Vasc. Med. 2009, 14, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Phillips, N.A.; Mate-Kole, C.C. Cognitive deficits in peripheral vascular disease. A comparison of mild stroke patients and normal control subjects. Stroke J. Cereb. Circ. 1997, 28, 777–784. [Google Scholar] [CrossRef]
- Rahimi, J.; Kovacs, G.G. Prevalence of mixed pathologies in the aging brain. Alzheimers Res. Ther. 2014, 6, 82. [Google Scholar] [CrossRef]
- Pluta, R.; Ulamek, M.; Jablonski, M. Alzheimer’s mechanisms in ischemic brain degeneration. Anat. Rec. (Hoboken) 2009, 292, 1863–1881. [Google Scholar] [CrossRef]
- Marchant, N.L.; Reed, B.R.; Sanossian, N.; Madison, C.M.; Kriger, S.; Dhada, R.; Mack, W.J.; DeCarli, C.; Weiner, M.W.; Mungas, D.M.; et al. The aging brain and cognition: Contribution of vascular injury and abeta to mild cognitive dysfunction. JAMA Neurol. 2013, 70, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Arvanitakis, Z.; Bang, W.; Bennett, D.A. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007, 69, 2197–2204. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Guo, Q.; Inoue, T.; Polito, V.A.; Tabuchi, K.; Hammer, R.E.; Pautler, R.G.; Taffet, G.E.; Zheng, H. Vascular and parenchymal amyloid pathology in an Alzheimer disease knock-in mouse model: Interplay with cerebral blood flow. Mol. Neurodegener. 2014, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, S.; Reed, B.R.; Madison, C.M.; Wirth, M.; Marchant, N.L.; Kriger, S.; Mack, W.J.; Sanossian, N.; DeCarli, C.; Chui, H.C.; et al. Vascular risk and Aβ interact to reduce cortical thickness in AD vulnerable brain regions. Neurology 2014, 83, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.W.; Parker, D.E.; Montgomery, P.S.; Sosnowska, D.; Casanegra, A.I.; Ungvari, Z.; Csiszar, A.; Sonntag, W.E. Greater endothelial apoptosis and oxidative stress in patients with peripheral artery disease. Int. J. Vasc. Med. 2014, 2014, 160534. [Google Scholar] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552 Pt 2, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.T.; Lin, H.T.; Hwang, P.A.; Wang, S.T.; Hsieh, C.H.; Hwang, D.F. Taurine resumed neuronal differentiation in arsenite-treated N2a cells through reducing oxidative stress, endoplasmic reticulum stress, and mitochondrial dysfunction. Amino Acids 2015, 47, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Katayama, T.; Imaizumi, K.; Manabe, T.; Hitomi, J.; Kudo, T.; Tohyama, M. Induction of neuronal death by ER stress in Alzheimer’s disease. J. Chem. Neuroanat. 2004, 28, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Yenki, P.; Khodagholi, F.; Shaerzadeh, F. Inhibition of phosphorylation of JNK suppresses Aβ-induced ER stress and upregulates prosurvival mitochondrial proteins in rat hippocampus. J. Mol. Neurosci. 2013, 49, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.C.; Lu, F.J.; Huang, C.F.; Chen, W.K.; Liu, S.H.; Lin-Shiau, S.Y. The diabetogenic effects of the combination of humic acid and arsenic: In vitro and in vivo studies. Toxicol. Lett. 2007, 172, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.L.; Ho, H.Y.; Chiu, D.T.; Lu, F.J. Humic acid-mediated oxidative damages to human erythrocytes: A possible mechanism leading to anemia in Blackfoot disease. Free Radic. Biol. Med. 1999, 27, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.L.; Huang, P.J.; Chen, S.C.; Cho, H.J.; Kumar, K.J.; Lu, F.J.; Chen, C.S.; Chang, C.T.; Hseu, Y.C. Induction of macrophage cell-cycle arrest and apoptosis by humic acid. Environ. Mol. Mutagen. 2014, 55, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Yang, D.I.; Lin, T.K.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Roles of oxidative stress, apoptosis, PGC-1α and mitochondrial biogenesis in cerebral ischemia. Int. J. Mol. Sci. 2011, 12, 7199–7215. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Inhibitors of mitochondrial fission as a therapeutic strategy for diseases with oxidative stress and mitochondrial dysfunction. J. Alzheimers Dis. 2014, 40, 245–256. [Google Scholar] [PubMed]
- Aliev, G.; Priyadarshini, M.; Reddy, V.P.; Grieg, N.H.; Kaminsky, Y.; Cacabelos, R.; Ashraf, G.M.; Jabir, N.R.; Kamal, M.A.; Nikolenko, V.N.; et al. Oxidative stress mediated mitochondrial and vascular lesions as markers in the pathogenesis of Alzheimer disease. Curr. Med. Chem. 2014, 21, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Mitochondria and neurodegeneration. Novartis Found. Symp. 2007, 287, 183–192. [Google Scholar] [PubMed]
- Austin, S.; St-Pierre, J. PGC1α and mitochondrial metabolism—Emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125 Pt 21, 4963–4971. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.C.; Keeney, P.M.; Algarzae, N.K.; Ladd, A.C.; Thomas, R.R.; Bennett, J.P., Jr. Mitochondrial DNA copy numbers in pyramidal neurons are decreased and mitochondrial biogenesis transcriptome signaling is disrupted in Alzheimer’s disease hippocampi. J. Alzheimers Dis. 2014, 40, 319–330. [Google Scholar] [PubMed]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1α expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [PubMed]
- Procaccio, V.; Bris, C.; Chao de la Barca, J.M.; Oca, F.; Chevrollier, A.; Amati-Bonneau, P.; Bonneau, D.; Reynier, P. Perspectives of drug-based neuroprotection targeting mitochondria. Revue Neurol. 2014, 170, 390–400. [Google Scholar] [CrossRef] [Green Version]
- Diao, L.; Marshall, A.H.; Dai, X.; Bogdanovic, E.; Abdullahi, A.; Amini-Nik, S.; Jeschke, M.G. Burn plus lipopolysaccharide augments endoplasmic reticulum stress and NLRP3 inflammasome activation and reduces PGC-1α in liver. Shock (Augusta) 2014, 41, 138–144. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Wareski, P.; Vaarmann, A.; Choubey, V.; Safiulina, D.; Liiv, J.; Kuum, M.; Kaasik, A. PGC-1α and PGC-1β regulate mitochondrial density in neurons. J. Biol. Chem. 2009, 284, 21379–21385. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.S.; Fonseca-Kelly, Z.; Callinan, C.; Zuo, L.; Sachdeva, M.M.; Shindler, K.S. SIRT1 activating compounds reduce oxidative stress and prevent cell death in neuronal cells. Front. Cell. Neurosci. 2012, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Zaccone, C.; Soler-Rovira, P.; Plaza, C.; Cocozza, C.; Miano, T.M. Variability in As, Ca, Cr, K, Mn, Sr, and Ti concentrations among humic acids isolated from peat using NaOH, Na4P2O7 and NaOH+Na4P2O7 solutions. J. Hazard. Mater. 2009, 167, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Zaccone, C.; Sanei, H.; Outridge, P.M.; Miano, T.M. Studying the humification degree and evolution of peat down a Holocene bog profile (Inuvik, NW, Canada): A petrological and chemical perspective. Org. Geochem. 2011, 42, 399–408. [Google Scholar] [CrossRef]
- Topal, A.; Atamanalp, M.; Alak, G.; Oruç, E.; Kocaman, E.M.; Sağlam, Y.S. Effect of humic acid on the brain tissue of brown trout treated with cadmium. Int. J. Fish. Aquat. Stud. 2014, 1, 18–21. [Google Scholar]
- Kurapati, K.R.; Samikkannu, T.; Atluri, V.S.; Kaftanovskaya, E.; Yndart, A.; Nair, M.P. β-Amyloid1–42, HIV-1Ba-L (clade B) infection and drugs of abuse induced degeneration in human neuronal cells and protective effects of ashwagandha (Withania somnifera) and its constituent Withanolide A. PLoS ONE 2014, 9, e112818. [Google Scholar]
- Hseu, Y.C.; Yang, H.L. The effects of humic acid–arsenate complexes on human red blood cells. Environ. Res. 2002, 89, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Selvey, S.; Thompson, E.W.; Matthaei, K.; Lea, R.A.; Irving, M.G.; Griffiths, L.R. Beta-actin—An unsuitable internal control for RT-PCR. Mol. Cell. Probes 2001, 15, 307–311. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.-H.; Lu, F.-J.; Hung, H.-C.; Liu, G.-Y.; Lai, T.-J.; Lin, C.-L. Humic Acid Increases Amyloid β-Induced Cytotoxicity by Induction of ER Stress in Human SK-N-MC Neuronal Cells. Int. J. Mol. Sci. 2015, 16, 10426-10442. https://doi.org/10.3390/ijms160510426
Li H-H, Lu F-J, Hung H-C, Liu G-Y, Lai T-J, Lin C-L. Humic Acid Increases Amyloid β-Induced Cytotoxicity by Induction of ER Stress in Human SK-N-MC Neuronal Cells. International Journal of Molecular Sciences. 2015; 16(5):10426-10442. https://doi.org/10.3390/ijms160510426
Chicago/Turabian StyleLi, Hsin-Hua, Fung-Jou Lu, Hui-Chih Hung, Guang-Yaw Liu, Te-Jen Lai, and Chih-Li Lin. 2015. "Humic Acid Increases Amyloid β-Induced Cytotoxicity by Induction of ER Stress in Human SK-N-MC Neuronal Cells" International Journal of Molecular Sciences 16, no. 5: 10426-10442. https://doi.org/10.3390/ijms160510426
APA StyleLi, H. -H., Lu, F. -J., Hung, H. -C., Liu, G. -Y., Lai, T. -J., & Lin, C. -L. (2015). Humic Acid Increases Amyloid β-Induced Cytotoxicity by Induction of ER Stress in Human SK-N-MC Neuronal Cells. International Journal of Molecular Sciences, 16(5), 10426-10442. https://doi.org/10.3390/ijms160510426