
Global Proteomic Analysis of Brain Tissues in Transient Ischemia Brain Damage in Rats



Abstract
:1. Introduction
2. Results and Discussion
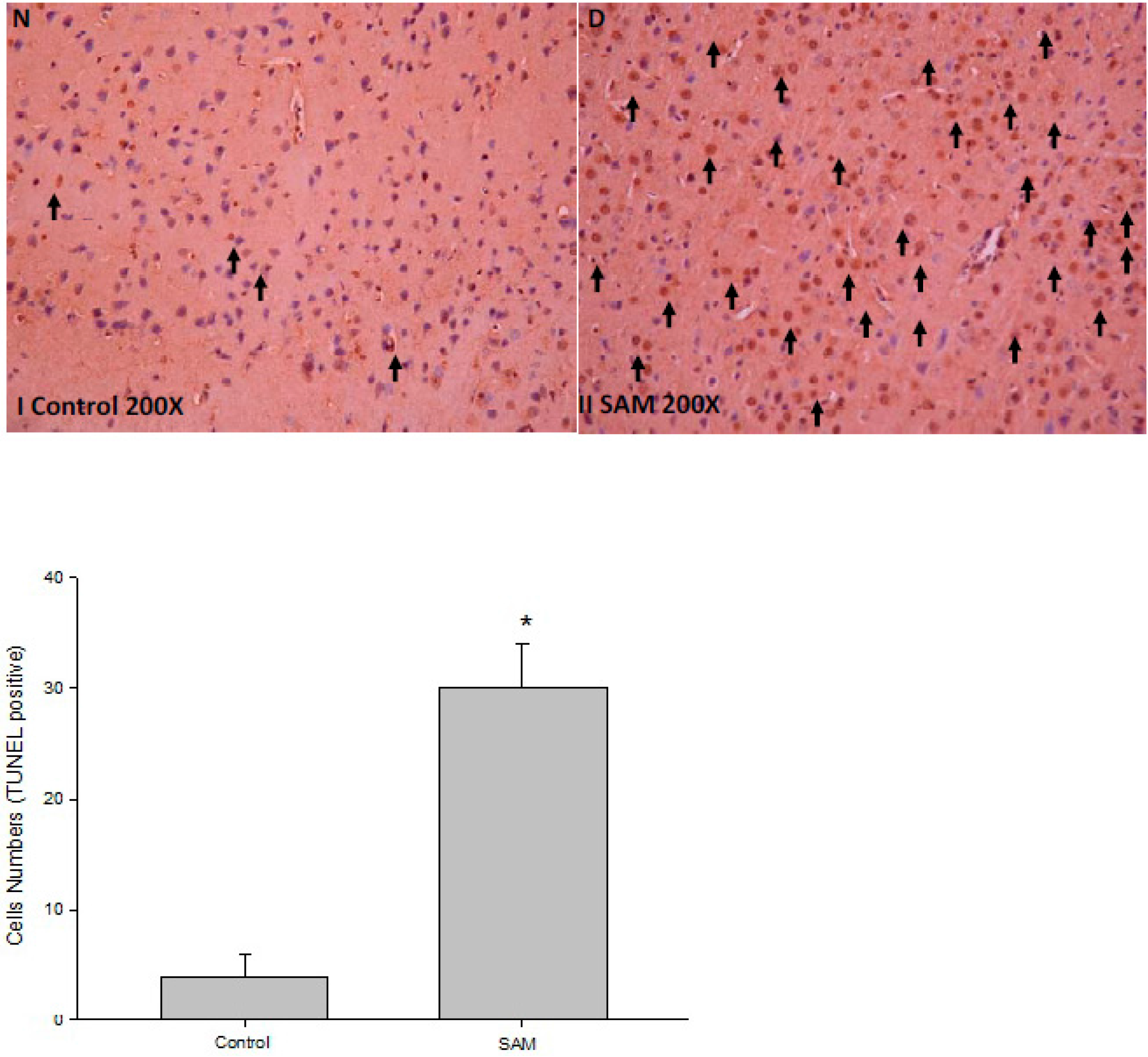
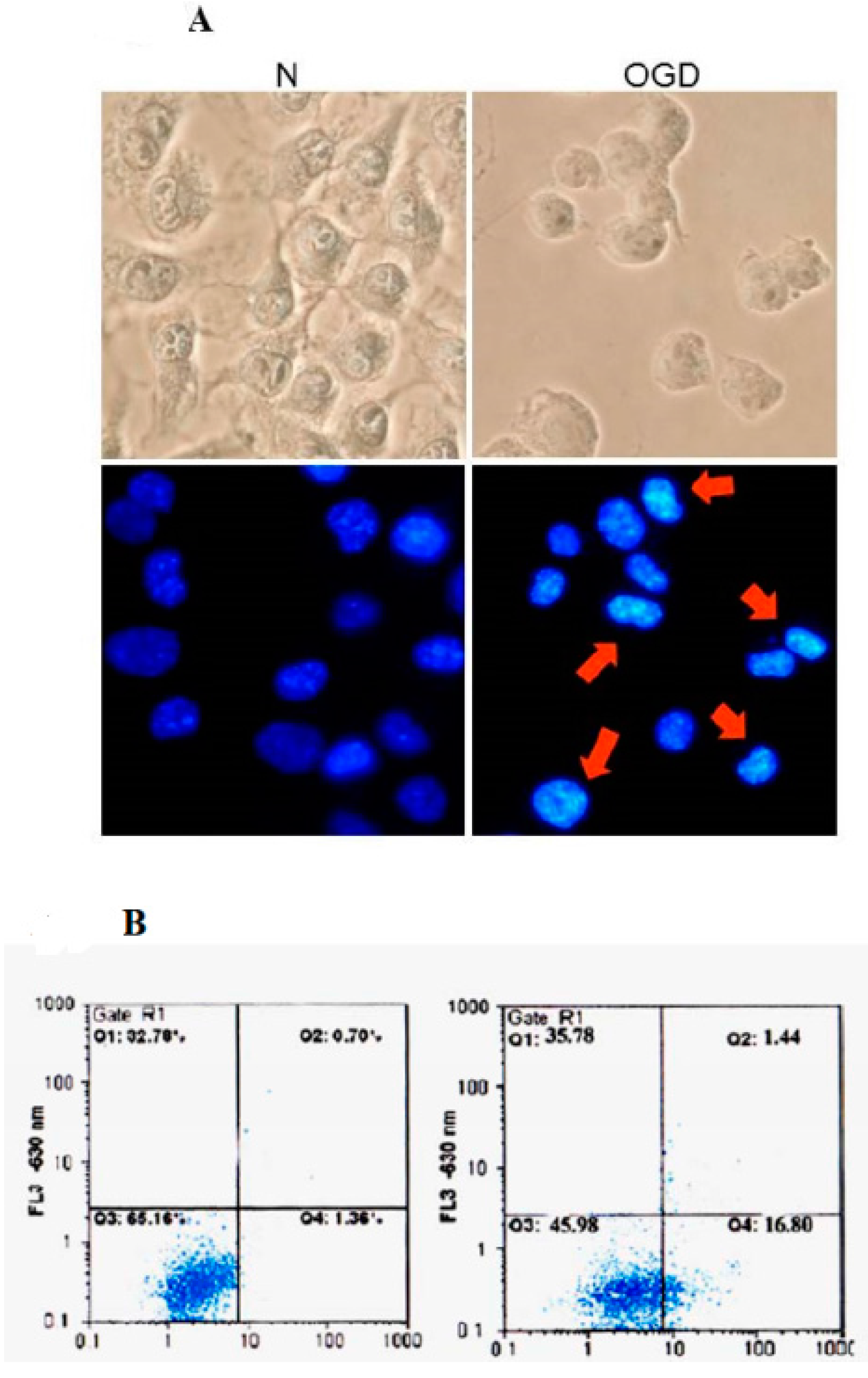
2.1. Cytotoxic Effect of Neuron Cells in Ischemic Rats

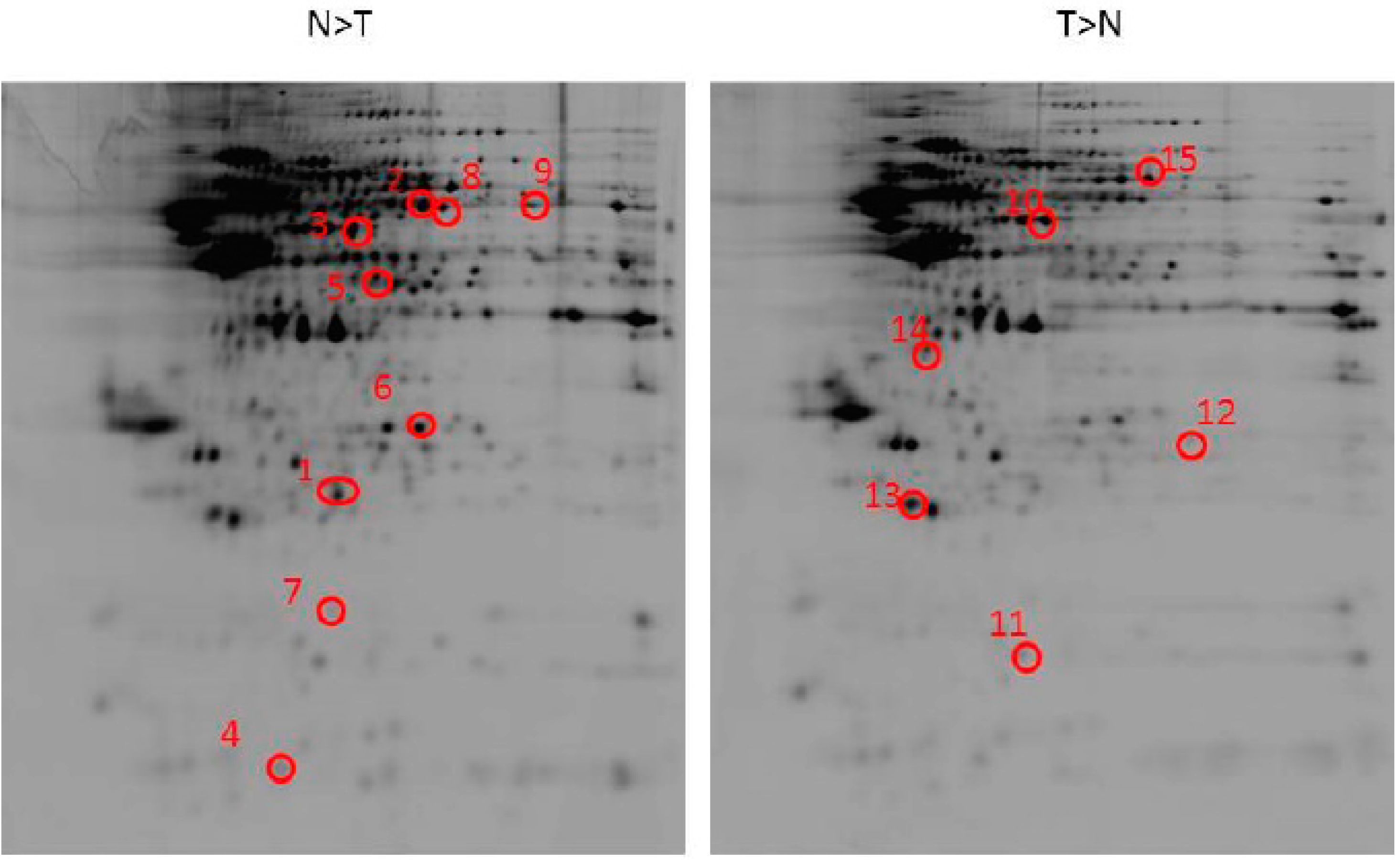
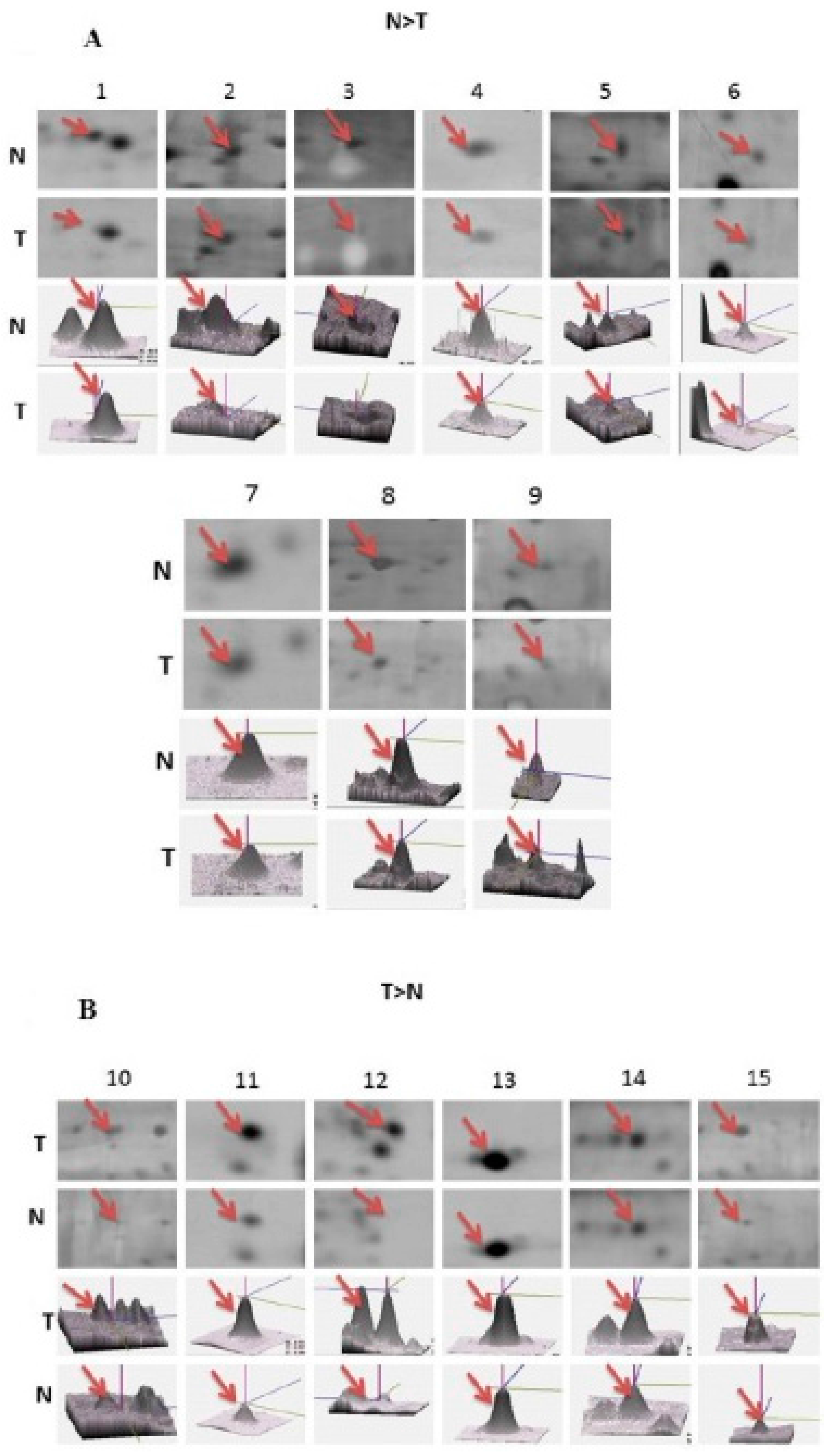
2.2. Identification of Differentially Expressed Proteins after Ischemia Reperfusion Brain Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spot | Protein Name | Mr/PI | Accession No. | MASCOT Score | Matched Peptides |
|---|---|---|---|---|---|
| 1. | Catechol O-methyltransferase | 29/5.3 | COMT_RAT | 637 | 47 |
| 2. | Protein disulfide-isomerase A3 | 56/8.8 | PDIA3_RAT | 108 | 21 |
| 3. | Guanine deaminase | 50/6.0 | GUAD_RAT | 58 | 15 |
| 4. | Cystic fibrosis transmembrane conductance regulator | 167/9.5 | CFTR_RAT | 89 | 10 |
| 5. | Cathepsin D | 44/6.8 | CATD_RAT | 375 | 21 |
| 6. | Guanidinoacetate N-methyltransferase | 26/5.6 | GAMT_RAT | 437 | 30 |
| 7. | d-dopachrome decarboxylase | 13/6.1 | DOPD_RAT | 332 | 27 |
| 9. | d-3-phosphoglycerate dehydrogenase | 26/5.6 | SERA_RAT | 437 | 30 |
| 10. | Dihydropyrimidinase-related protein 2 | 62/6.4 | DPYL2_RAT | 59 | 14 |
| 11. | Histidine triad nucleotide-binding protein 1 | 13/5.5 | HINT1_RAT | 63 | 9 |
| 12. | Protein-l-isoaspartate (d-aspartate)O-methyltransferase | 24/7.8 | PIMT_RAT | 483 | 27 |
| 13. | Beta-synuclein | 14/4.3 | SYUB_RAT | 714 | 105 |
| 14. | Calretinin | 31/9.6 | CALB2_RAT | 73 | 13 |
| 15. | Neurexin-3-alpha | 173/6.1 | NRX3A_RAT | 94 | 16 |


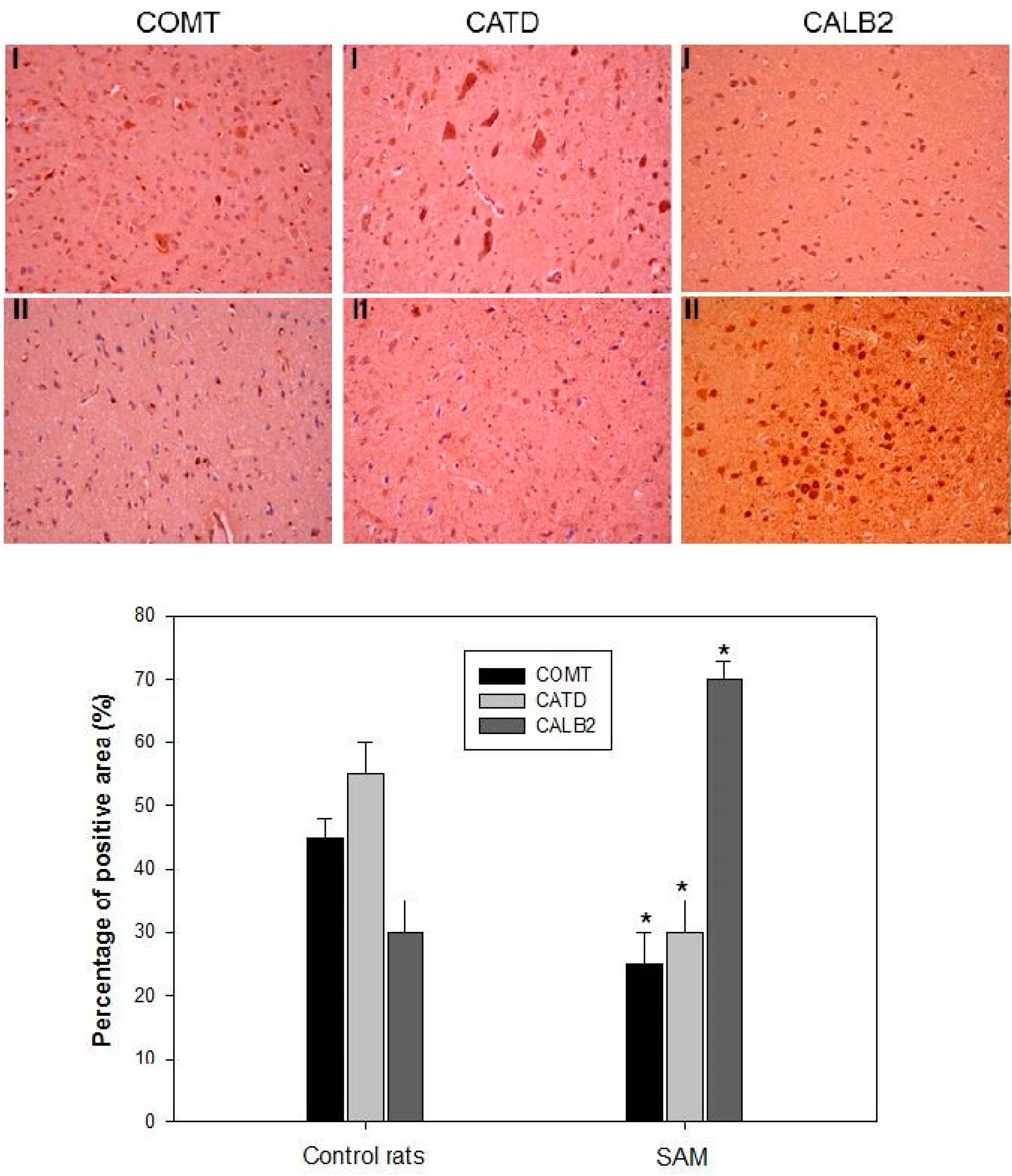
2.3. Validation of Differentially Displayed Proteins on Brain Neuron Histopathological Changes

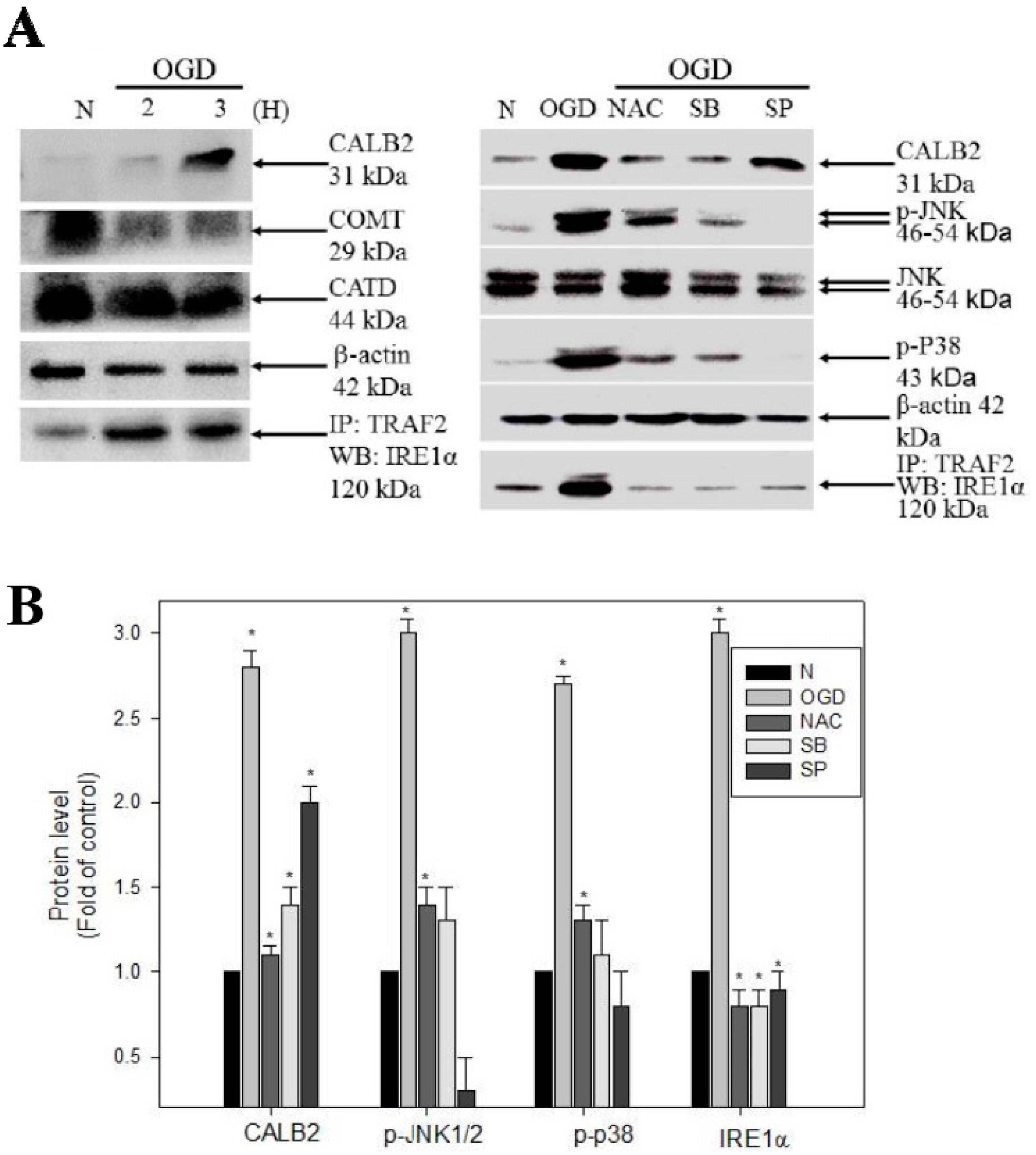
2.4. Effects of OGD Treatment on the Up-Regulation of CALB2, the ER Stress-Signaling Mechanisms and on ROS-Related Apoptosis
| % of Cell Death | SOD Activity (%) | |
|---|---|---|
| Control | 2 | 100 |
| OGD | 21 | 35 |
| OGD-SB | 10 | 77 |
| OGD-SP | 8 | 80 |
| OGD-NAC | 5 | 88 |

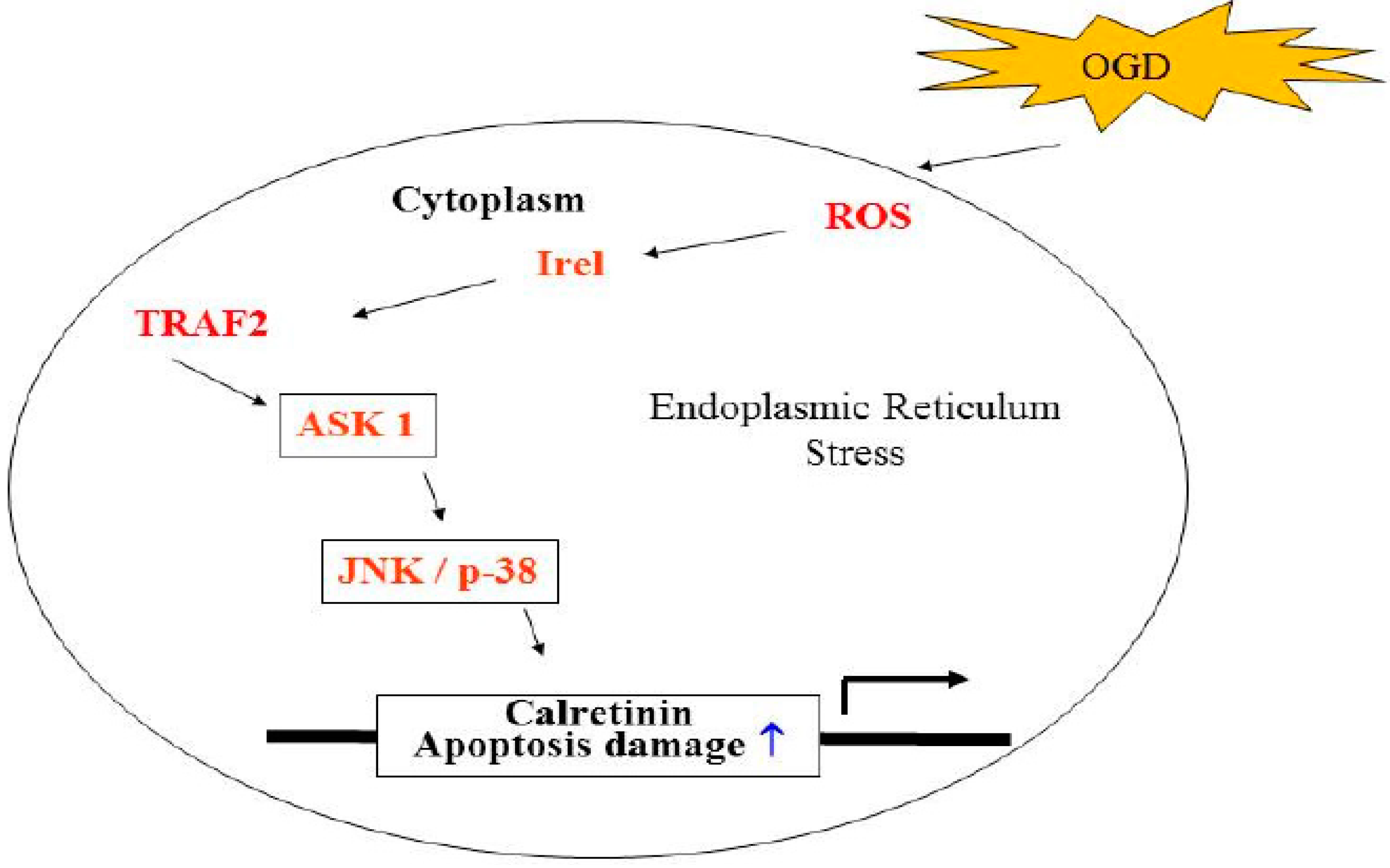
2.5. ROS, IRE1-alpha/TRAF2, JNK1/2/, and p38 MAPK Triggering Pathways Are Involved in the Regulation of OGD-Induced CALB2 Expression in Neuron Cells


3. Experimental Section
3.1. Induction of Ischemia Reperfusion Brain Injury
3.2. Chemicals and Reagents
3.3. Analysis of Brain Proteomic Profiles in Transient Ischemia Rats Using Two-Dimensional Protein Electrophoresis
3.4. In-Gel Digestion and Identification of Peptide Fingerprints with Serum Proteins Using MALDI-TOF
3.5. Immunohistochemistry
3.6. Oxygen Glucose Deprivation (OGD) of Ischemia
3.7. Statistical Analyses
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Towfighi, A.; Saver, J.L. Stroke declines from third to fourth leading cause of death in the United States: Historical perspective and challenges ahead. Stroke 2011, 42, 2351–2355. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wang, X.; Xu, F.; Bahr, B.A.; Shibata, M.; Uchiyama, Y.; Hagberg, H.; Blomgren, K. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ. 2005, 12, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [PubMed]
- Meisel, C.; Schwab, J.M.; Prass, K.; Meisel, A.; Dirnagl, U. Central nervous system injury-induced immune deficiency syndrome. Nat. Rev. Neurosci. 2005, 6, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Urra, X.; Cervera, A.; Villamor, N.; Planas, A.M.; Chamorro, A. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience 2009, 158, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Blomgren, K.; Leist, M.; Groc, L. Pathological apoptosis in the developing brain. Apoptosis 2007, 12, 993–1010. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Qiu, L.; Wang, X.; Hallin, U.; Candé, C.; Kroemer, G.; Hagberg, H.; Blomgren, K. Involvement of apoptosis-inducing factor in neuronal death after hypoxia-ischemia in the neonatal rat brain. J. Neurochem. 2003, 86, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Carden, D.L.; Granger, D.N. Pathophysiology of ischaemia-reperfusion injury. J. Pathol. 2000, 190, 255–266. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [PubMed]
- Todd, D.J.; Lee, A.H.; Glimcher, L.H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008, 8, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Bredesen, D.E. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr. Opin. Cell Biol. 2004, 16, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Benbrook, D.M.; Long, A. Integration of autophagy, proteasomal degradation, unfolded protein response and apoptosis. Exp. Oncol. 2012, 34, 286–297. [Google Scholar] [PubMed]
- Chen, J.H.; Kuo, H.C.; Lee, K.F.; Tsai, T.H. Magnolol protects neurons against ischemia injury via the downregulation of p38/MAPK, CHOP and nitrotyrosine. Toxicol. Appl. Pharmacol. 2014, 279, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Badiola, N.; Penas, C.; Miñano-Molina, A.; Barneda-Zahonero, B.; Fadó, R.; Sánchez-Opazo, G.; Comella, J.X.; Sabriá, J.; Zhu, C.; Blomgren, K.; et al. Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis. 2011, 2, e149. [Google Scholar] [CrossRef] [PubMed]
- Malagelada, C.; Xifró, X.; Miñano, A.; Sabriá, J.; Rodríguez-Alvarez, J. Contribution of caspase-mediated apoptosis to the cell death caused by oxygen-glucose deprivation in cortical cell cultures. Neurobiol. Dis. 2005, 20, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Maier, P.J.; Zemoura, K.; Acuña, M.A.; Yévenes, G.E.; Zeilhofer, H.U.; Benke, D. Ischemia-like oxygen and glucose deprivation mediates down-regulation of cell surface γ-aminobutyric acidB receptors via the endoplasmic reticulum (ER) stress-induced transcription factor CCAAT/enhancer-binding protein (C/EBP)-homologous protein (CHOP). J. Biol. Chem. 2014, 289, 12896–12907. [Google Scholar] [PubMed]
- Simi, A.; Ingelman-Sundberg, M.; Tindberg, N. Neuroprotective agent chlomethiazole attenuates c-fos, c-jun, and AP-1 activation through inhibition of p38 MAP kinase. J. Cereb. Blood Flow. MeTable 2000, 20, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.P.; Connell, J.; Blackstone, K.; Ringler, S.L. Loss of calcium and increased apoptosis within the same neuron. Brain Res. 2007, 1128, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Camp, A.J.; Wijesinghe, R. Calretinin: Modulator of neuronal excitability. Int. J. Biochem. Cell Biol. 2009, 41, 2118–2121. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.; Talwar, P.; Parimisetty, A.; Lefebvre d’Hellencourt, C.; Ravanan, P. A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Front. Cell Neurosci. 2014, 8, 213. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.R.; Keller, J.N. Oxidative stress and cerebral endothelial cells: Regulation of the blood-brain-barrier and antioxidant based interventions. Biochim. Biophys. Acta 2012, 1822, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Song, J.; Yuan, P.; Tian, Q.; Ji, Y.; Ren-Patterson, R.; Liu, G.; Sei, Y.; Weinberger, D.R. Orientation and cellular distribution of membrane-bound catechol-O-methyltransferase in cortical neurons: Implications for drug development. J. Biol. Chem. 2011, 286, 34752–34760. [Google Scholar] [CrossRef] [PubMed]
- Okada, R.; Wu, Z.; Zhu, A.; Ni, J.; Zhang, J.; Yoshimine, Y.; Peters, C.; Saftig, P.; Nakanishi, H. Cathepsin D deficiency induces oxidative damage in brain pericytes and impairs the blood-brain barrier. Mol. Cell. Neurosci. 2015, 64, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Rea, H.C.; Wiktorowicz, J.E.; Perez-Polo, J.R. Proteomic analysis of hypoxia/ischemia-induced alteration of cortical development and dopamine neurotransmission in neonatal rat. J. Proteome Res. 2006, 5, 2396–2404. [Google Scholar] [CrossRef] [PubMed]
- Bearzatto, B.; Servais, L.; Roussel, C.; Gall, D.; Baba-Aïssa, F.; Schurmans, S.; de Kerchove d’Exaerde, A.; Cheron, G.; Schiffmann, S.N. Targeted calretinin expression in granule cells of calretinin-null mice restores normal cerebellar functions. FASEB J. 2006, 20, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Schurmans, S.; Schiffmann, S.N.; Gurden, H.; Lemaire, M.; Lipp, H.P.; Schwam, V.; Pochet, R.; Imperato, A.; Böhme, G.A.; Parmentier, M. Impaired long-term potentiation induction in dentate gyrus of calretinin-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 10415–10420. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Lust, W.D.; Mrsulja, B.B.; Mrsulja, B.J.; Passonneau, J.V.; Klatzo, I. Putative neurotransmitters and cyclic nucleotides in prolonged ischemia of the cerebral cortex. Brain Res. 1975, 98, 394–399. [Google Scholar] [CrossRef]
- Calvert, J.; Zhang, J. Pathophysiology of an hypoxic-ischemic insult during the perinatal period. Neurol. Res. 2005, 27, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.L.; Bendek, G.; Dahlgren, N.; Rosén, I.; Wieloch, T.; Siesjö, B.K. Models for studying long-term recovery following forebrain ischemia in the rat. 2. A 2-vessel occlusion model. Acta Neurol. Scand. 1984, 69, 385–401. [Google Scholar] [CrossRef] [PubMed]
- Tseng, T.H.; Shen, C.H.; Huang, W.S.; Chen, C.N.; Liang, W.H.; Lin, T.H.; Kuo, H.C. Activation of neutral-sphingomyelinase, MAPKs, and p75 NTR-mediating caffeic acid phenethyl ester-induced apoptosis in C6 glioma cells. J. Biomed. Sci. 2014, 21, 61. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.F.; Chen, J.H.; Teng, C.C.; Shen, C.H.; Hsieh, M.C.; Lu, C.C.; Lee, K.C.; Lee, L.Y.; Chen, W.P.; Chen, C.C.; et al. Protective Effects of Hericium erinaceus Mycelium and Its Isolated Erinacine A against Ischemia-Injury-Induced Neuronal Cell Death via the Inhibition of iNOS/p38 MAPK and Nitrotyrosine. Int. J. Mol. Sci. 2014, 15, 15073–15089. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.J.; Kuo, H.C.; Lee, K.F.; Tsai, T.H. Glycyrrhizin represses total parenteral nutrition-associated acute liver injury in rats by suppressing endoplasmic reticulum stress. Int. J. Mol. Sci. 2013, 14, 12563–12580. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.C.; Chiu, C.C.; Chang, W.C.; Sheen, J.M.; Ou, C.Y.; Kuo, H.C.; Chen, R.F.; Hsu, T.Y.; Chang, J.C.; Hsaio, C.C.; et al. Use of proteomic differential displays to assess functional discrepancies and adjustments of human bone marrow- and Wharton jelly-derived mesenchymal stem cells. J. Proteome Res. 2011, 10, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.S.; Kuo, Y.H.; Chin, C.C.; Wang, J.Y.; Yu, H.R.; Sheen, J.M.; Tung, S.Y.; Shen, C.H.; Chen, T.C.; Sung, M.L.; et al. Proteomic analysis of the effects of baicalein on colorectal cancer cells. Proteomics 2012, 12, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.H.; Tung, S.Y.; Huang, W.S.; Lu, C.C.; Lee, K.C.; Hsieh, Y.Y.; Chang, P.J.; Liang, H.F.; Chen, J.H.; Lin, T.H.; et al. Exploring the effects of tert-butylhydroperoxide induced liver injury using proteomic approach. Toxicology 2014, 316, 61–70. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.-H.; Kuo, H.-C.; Lee, K.-F.; Tsai, T.-H. Global Proteomic Analysis of Brain Tissues in Transient Ischemia Brain Damage in Rats. Int. J. Mol. Sci. 2015, 16, 11873-11891. https://doi.org/10.3390/ijms160611873
Chen J-H, Kuo H-C, Lee K-F, Tsai T-H. Global Proteomic Analysis of Brain Tissues in Transient Ischemia Brain Damage in Rats. International Journal of Molecular Sciences. 2015; 16(6):11873-11891. https://doi.org/10.3390/ijms160611873
Chicago/Turabian StyleChen, Jiann-Hwa, Hsing-Chun Kuo, Kam-Fai Lee, and Tung-Hu Tsai. 2015. "Global Proteomic Analysis of Brain Tissues in Transient Ischemia Brain Damage in Rats" International Journal of Molecular Sciences 16, no. 6: 11873-11891. https://doi.org/10.3390/ijms160611873
APA StyleChen, J. -H., Kuo, H. -C., Lee, K. -F., & Tsai, T. -H. (2015). Global Proteomic Analysis of Brain Tissues in Transient Ischemia Brain Damage in Rats. International Journal of Molecular Sciences, 16(6), 11873-11891. https://doi.org/10.3390/ijms160611873