
Baicalin Protects Mice from Aristolochic Acid I-Induced Kidney Injury by Induction of CYP1A through the Aromatic Hydrocarbon Receptor

Abstract
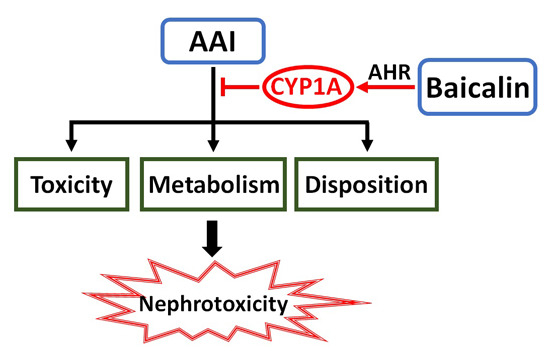
:
1. Introduction
2. Results and Discussion
2.1. Results
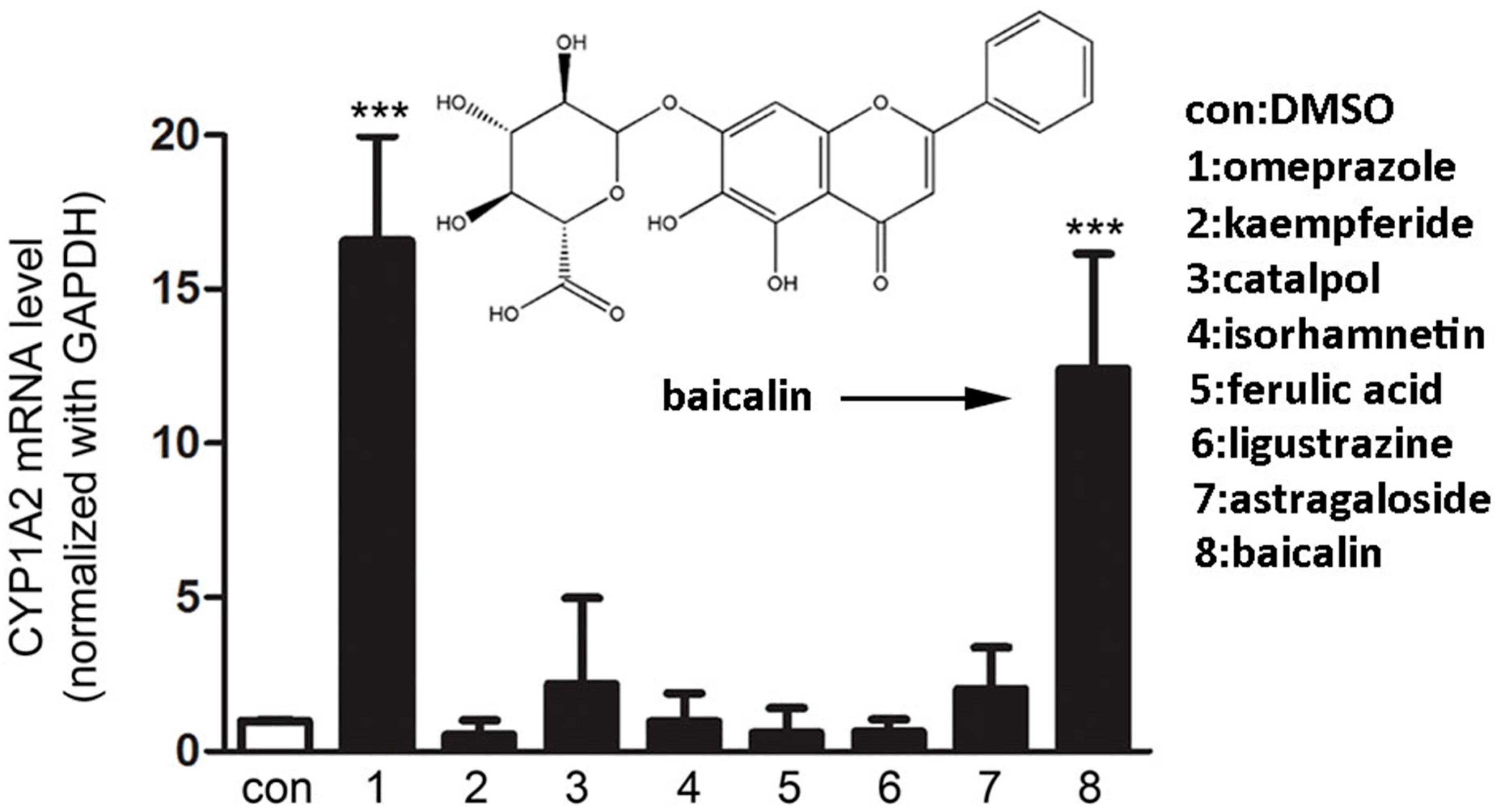
2.1.1. Screening of Herbal Compounds with CYP1A2 Induction Assays

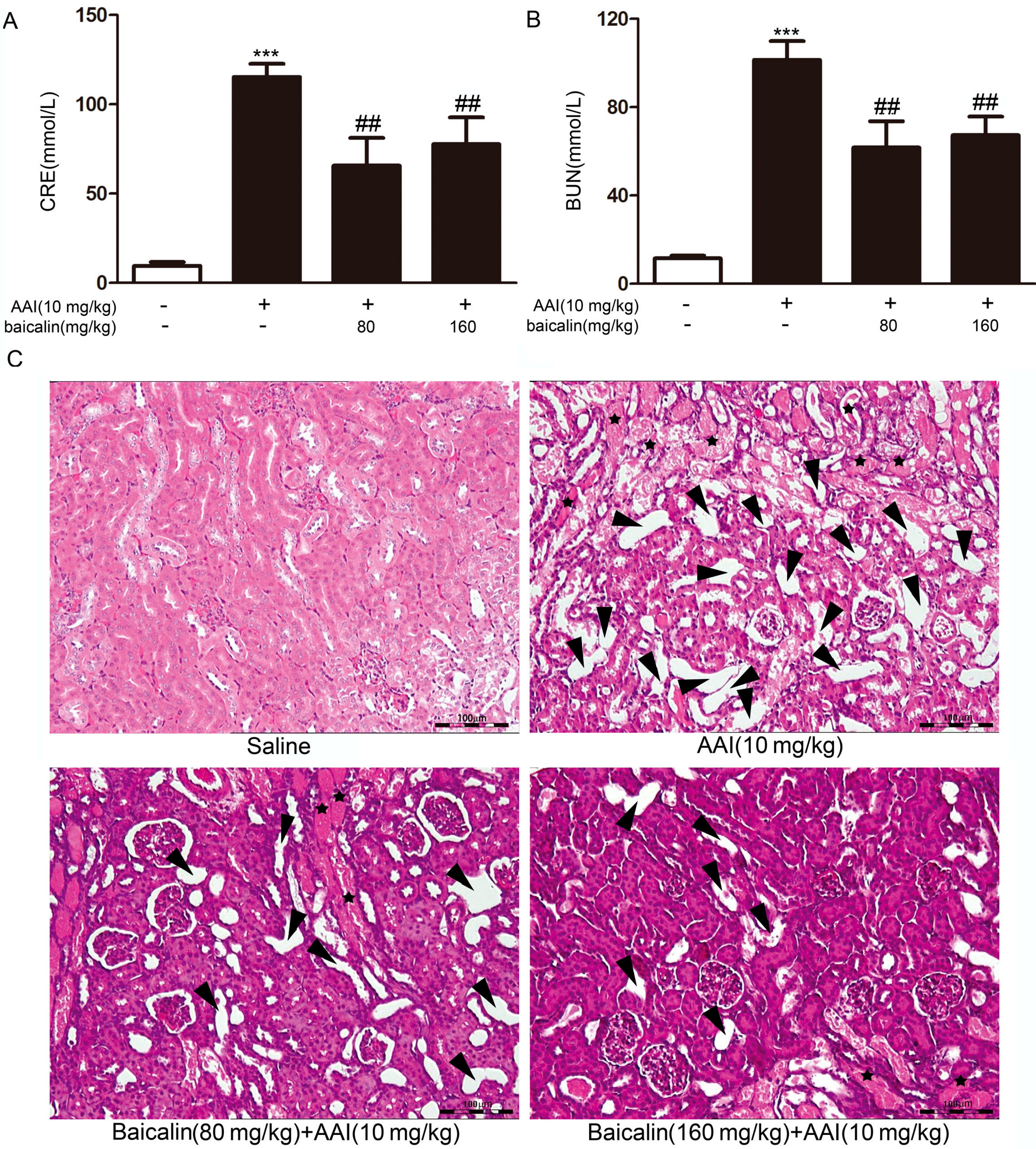
2.1.2. Effects of Baicalin on Aristolochic Acid I (AAI)-Induced Renal Damage

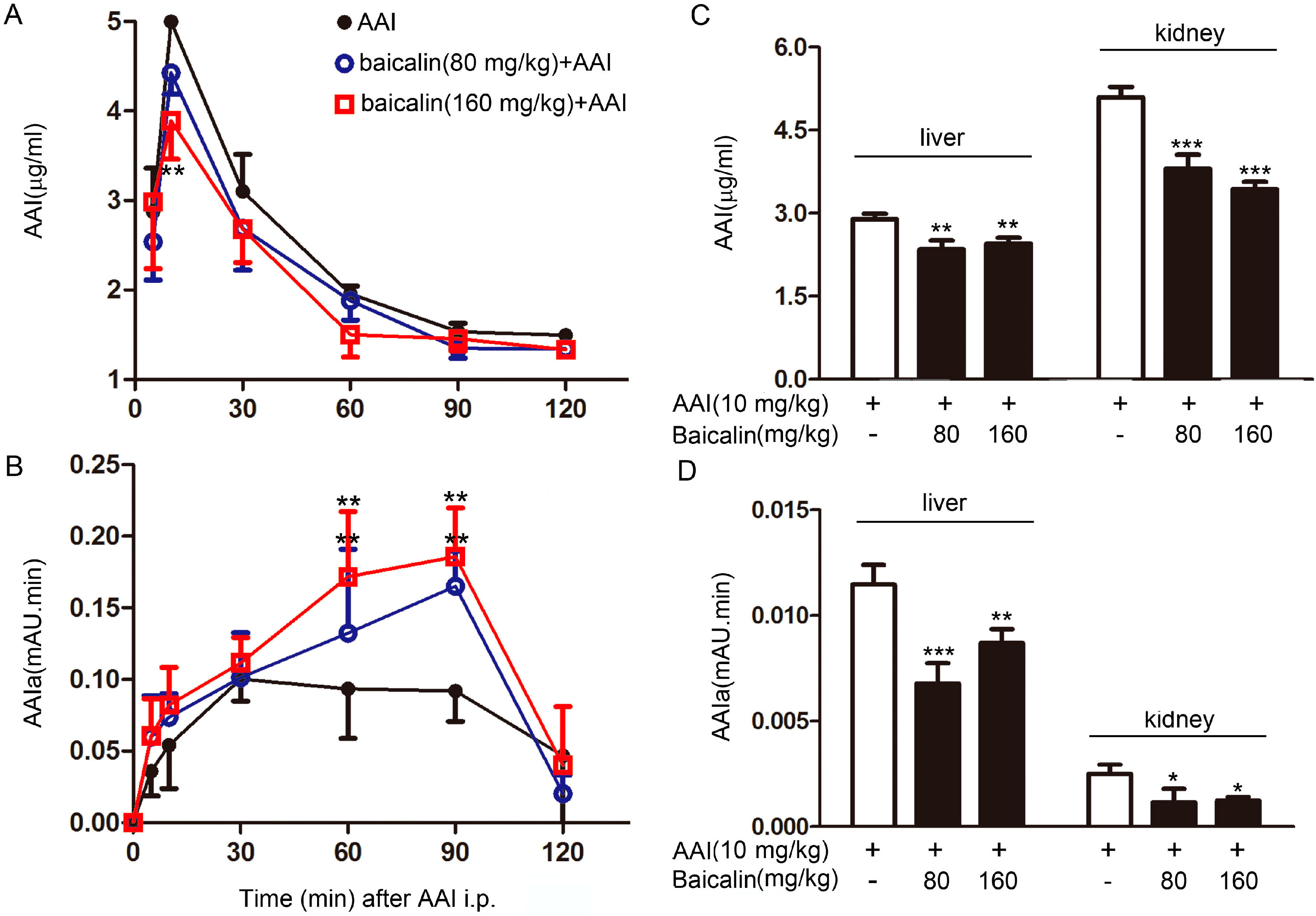
2.1.3. Effects of Baicalin on AAI metabolism in the Plasma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Cmax (μg/mL) | Tmax (min) | AUC (min·μg/mL) | t1/2 (min) |
|---|---|---|---|---|
| AAI | 4.99 ± 0.41 | 10.00 ± 0 | 844.96 ± 40.12 | 78.68 ± 12.88 |
| Baicalin (80 mg/kg) + AAI | 4.43 ± 0.24 | 10.00 ± 0 | 759.39 ± 23.26 ** | 77.96 ± 6.84 |
| Baicalin (160 mg/kg) + AAI | 3.89 ± 0.43 | 9 ± 2.24 | 710.07 ± 33.95 *** | 80.83 ± 3.02 |

2.1.4. Effects of Baicalin on AAI Distribution in Tissues
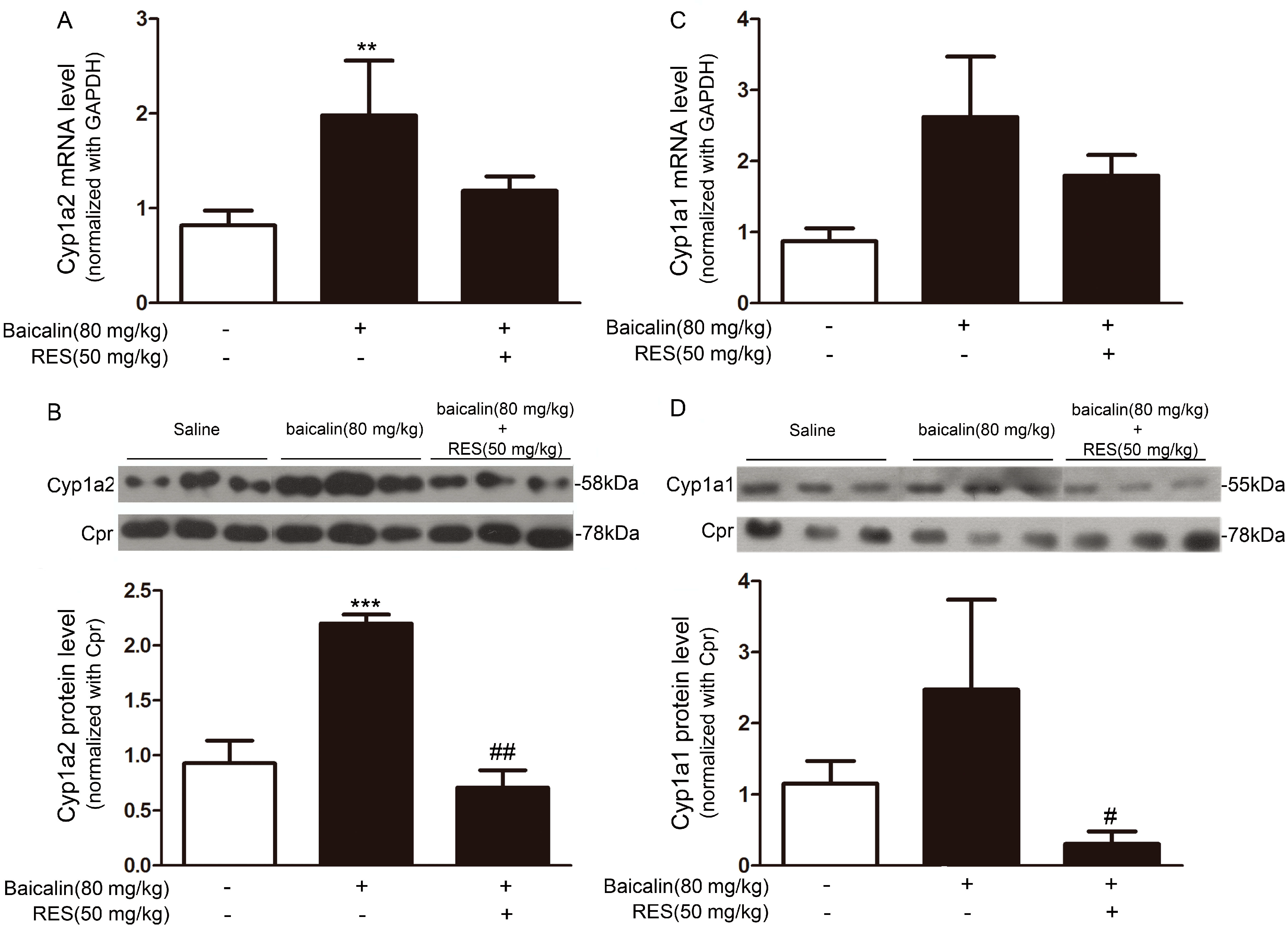
2.1.5. Mechanism Underpinning the Protective Effect of Baicalin against AA Injury


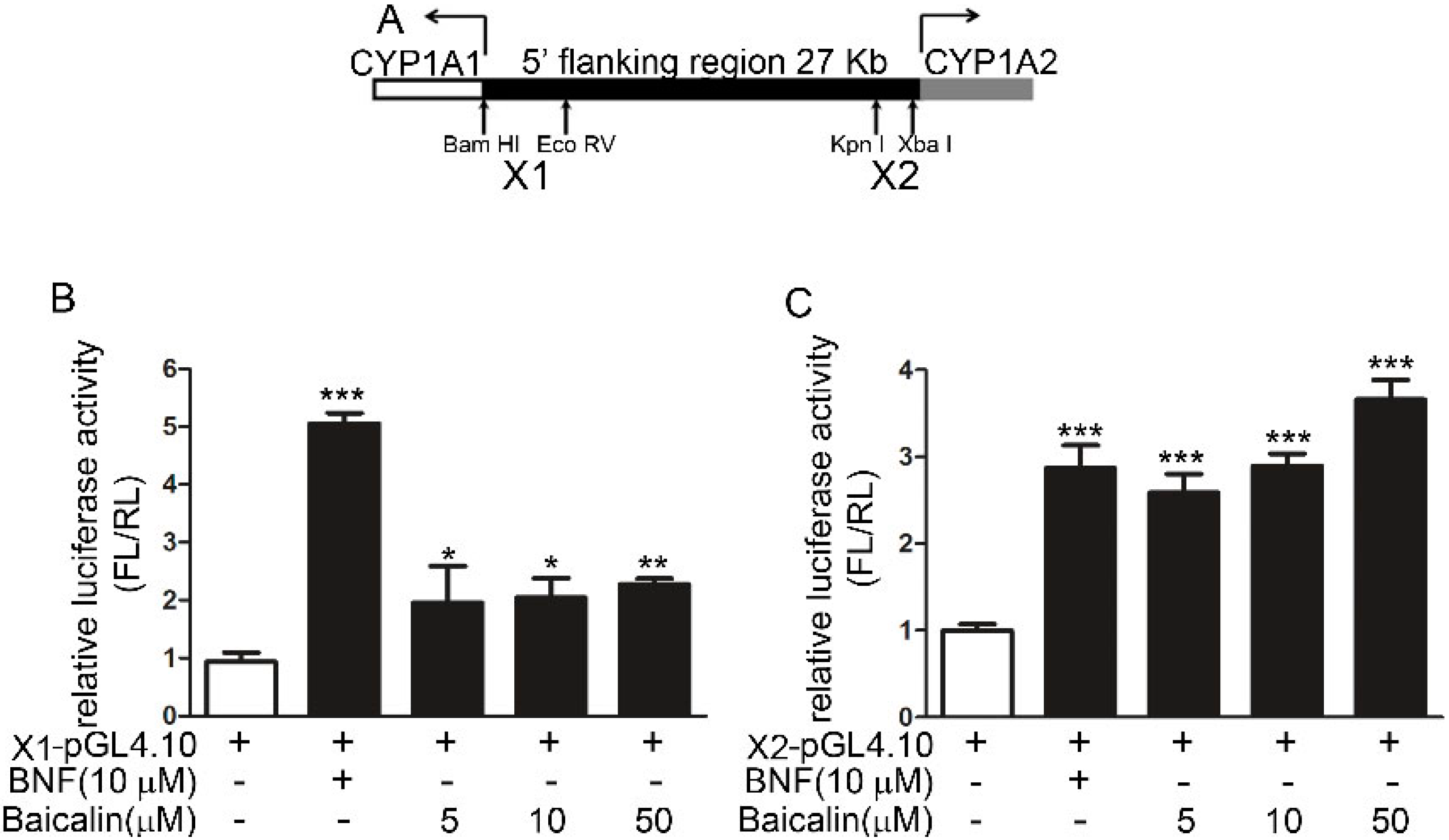
2.1.6. Role of AhR in Baicalin-Induced CYP1A Induction

2.2. Discussion
3. Materials and Methods
3.1. Chemicals
3.2. Cell Culture and Treatment
3.3. Animal Treatment

3.4. Histopathological Examination
3.5. Determination of AAI and Its Major Metabolites in the Blood, Liver, and Kidney
3.6. HPLC Analysis
3.7. Real-Time PCR Analysis
| Primer | Sequence (5′–3′) |
|---|---|
| CYP1A1 | Forward: CTTCCGACACTCTTCCTTCG |
| Reverse: ATAGCACCATCAGGGGTGAG | |
| CYP1A2 | Forward: GTCACCTCAGGGAATGCTGTG |
| Reverse: GTTGACAATCTTCTCCTGAGG | |
| GAPDH | Forward: GGTGGTCTCCTCTGACTTCAACA |
| Reverse: GTTGCTGTAGCCAAATTCGTTGT | |
| cyp1a1 | Forward: GACCCTTACAAGTATTTGGTCGT |
| Reverse: GGTATCCAGAGCCAGTAACCT | |
| cyp1a2 | Forward: CCAGGTGGTGGAATCGGTG |
| Reverse: TCTTAAACCTCTTGAGGGCCG | |
| gapdh | Forward: GGCTACACTGAGGACCAGGTT |
| Reverse: TGCTGTAGCCGTATTCATTGTC |
3.8. Western Blot Analysis
3.9. Plasmid Construction and Transfection
3.10. Luciferase Assay
3.11. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kuo, P.C.; Li, Y.C.; Wu, T.S. Chemical constituents and pharmacology of the Aristolochia (madou ling) species. J. Tradit. Complement. Med. 2012, 2, 249–266. [Google Scholar] [PubMed]
- Xue, X.; Xiao, Y.; Gong, L.; Guan, S.; Liu, Y.; Lu, H.; Qi, X.; Zhang, Y.; Li, Y.; Wu, X.; et al. Comparative 28-day repeated oral toxicity of Longdan Xieganwan, Akebia trifoliate (Thunb.) koidz., Akebia quinata (Thunb.) Decne. and Caulis aristolochiae manshuriensis in mice. J. Ethnopharmacol. 2008, 119, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Vanherweghem, J.L.; Depierreux, M.; Tielemans, C.; Abramowicz, D.; Dratwa, M.; Jadoul, M.; Richard, C.; Vandervelde, D.; Verbeelen, D.; Vanhaelen-Fastre, R.; et al. Rapidly progressive interstitial renal fibrosis in Young women: Association with slimming regimen including Chinese herbs. Lancet 1993, 341, 387–391. [Google Scholar] [CrossRef]
- De Broe, M.E. Chinese herbs nephropathy and Balkan endemic nephropathy: Toward a single entity, aristolochic acid nephropathy. Kidney Int. 2012, 81, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Jha, V. Herbal medicines and chronic kidney disease. Nephrology 2010, 15, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ge, M.; Xue, X.; Wang, C.; Wang, H.; Wu, X.; Li, L.; Liu, L.; Qi, X.; Zhang, Y.; et al. Hepatic cytochrome P450s metabolize aristolochic acid and reduce its kidney toxicity. Kidney Int. 2008, 73, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Xiao, Y.; Zhu, H.; Wang, H.; Liu, Y.; Xie, T.; Ren, J. Induction of P450 1A by 3-methylcholanthrene protects mice from aristolochic acid-I-induced acute renal injury. Nephrol. Dial. Transplant. 2008, 23, 3074–3081. [Google Scholar] [CrossRef] [PubMed]
- Stiborova, M.; Levova, K.; Barta, F.; Shi, Z.; Frei, E.; Schmeiser, H.H.; Nebert, D.W.; Phillips, D.H.; Arlt, V.M. Bioactivation versus detoxication of the urothelial carcinogen aristolochic acid I by human cytochrome P450 1A1 and 1A2. Toxicol. Sci. 2012, 125, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Xie, X.; Wu, M.; Li, C.; Gao, M.; Liu, M.; Qi, X.; Ren, J. Tanshinone I protects mice from aristolochic acid I-induced kidney injury by induction of CYP1A. Environ. Toxicol. Pharmacol. 2013, 36, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Moorthy, B.; Jiang, W.; Wang, L.; Chu, C.; Maturu, P. Molecular regulation of hepatic and Pulmonary cytochrome P4501A (CYP1A1) enzymes by 3-methylcholanthrene in mice: Role of CYP1A2. FASEB J. 2015, 29, 778.5. [Google Scholar]
- Couroucli, X.I.; Liang, Y.H.; Jiang, W.; Wang, L.; Barrios, R.; Yang, P.; Moorthy, B. Prenatal administration of the cytochrome P4501A inducer, β-naphthoflavone (BNF), attenuates hyperoxic lung injury in newborn mice: Implications for bronchopulmonary dysplasia (BPD) in premature infants. Toxicol. Appl. Pharmacol. 2011, 256, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Xue, X.; Wu, Y.F.; Xin, G.Z.; Qian, Y.; Xie, T.P.; Gong, L.K.; Ren, J. β-Naphthoflavone protects mice from aristolochic acid-I-induced acute kidney injury in a CYP1A dependent mechanism. Acta Pharmacol. Sin. 2009, 30, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
- Stiborova, M.F.E.; Hodek, P.; Wiessler, M.; Schmeiser, H.H. Human hepatic and renal microsomes, cytochromes P450 1A1/2, NADPH:cytochrome P450 reductase and prostaglandin H synthase mediate the formation of aristolochic acid-DNA adducts found in patients with urothelial cancer. Int. J. Cancer 2005, 113, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Hodek, P.; Krizkova, J.; Frei, E.; Singh, R.; Arlt, V.M.; Stiborova, M. Impact of β-naphthoflavone on genotoxicity of food-derived carcinogens. Neuro Endocrinol. Lett. 2011, 32, 25–34. [Google Scholar] [PubMed]
- Stengel, B. Chronic kidney disease and cancer: A troubling connection. J. Nephrol. 2010, 23, 253–262. [Google Scholar] [PubMed]
- Yang, H.Y.; Wang, J.D.; Lo, T.C.; Chen, P.C. Occupational kidney disease among Chinese herbalists exposed to herbs containing aristolochic acids. Occup. Environ. Med. 2011, 68, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Dalton, T.P.; Okey, A.B.; Gonzalez, F.J. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J. Biol. Chem. 2004, 279, 23847–23850. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Thomas, P. Effects of hypoxia exposure on hepatic cytochrome P450 1A (CYP1A) expression in Atlantic croaker: Molecular mechanisms of CYP1A down-regulation. PLoS ONE 2012, 7, e40825. [Google Scholar] [CrossRef] [PubMed]
- Stejskalova, L.; Pavek, P. The function of cytochrome P450 1A1 enzyme (CYP1A1) and aryl hydrocarbon receptor (AhR) in the placenta. Curr. Pharm. Biotechnol. 2011, 12, 715–730. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Levova, K.; Barta, F.; Shi, Z.; Evans, J.D.; Frei, E.; Schmeiser, H.H.; Nebert, D.W.; Phillips, D.H.; Stiborova, M. Role of P450 1A1 and P450 1A2 in bioactivation versus detoxication of the renal carcinogen aristolochic acid I: studies in Cyp1a1(−/−), Cyp1a2(−/−), and Cyp1a1/1a2(−/−) mice. Chem. Res. Toxicol. 2011, 24, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Levova, K.; Moserova, M.; Kotrbova, V.; Sulc, M.; Henderson, C.J.; Wolf, C.R.; Phillips, D.H.; Frei, E.; Schmeiser, H.H.; Mares, J.; et al. Role of cytochromes P450 1A1/2 in detoxication and activation of carcinogenic aristolochic acid I: Studies with the hepatic NADPH:cytochrome P450 reductase null (HRN) mouse model. Toxicol. Sci. 2011, 121, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Rosenquist, T.A.; Einolf, H.J.; Dickman, K.G.; Wang, L.; Smith, A.; Grollman, A.P. Cytochrome P450 1A2 detoxicates aristolochic acid in the mouse. Drug Metab. Dispos. 2010, 38, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Sato, W.; Suzuki, H.; Sasaki, T.; Kumagai, T.; Sakaguchi, S.; Mizugaki, M.; Miyairi, S.; Yamazoe, Y.; Nagata, K. Construction of a system that simultaneously evaluates CYP1A1 and CYP1A2 induction in a stable human-derived cell line using a dual reporter plasmid. Drug Metab. Pharmacokinet. 2010, 25, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Yoshinari, K.; Ueda, R.; Kusano, K.; Yoshimura, T.; Nagata, K.; Yamazoe, Y. Omeprazole transactivates human CYP1A1 and CYP1A2 expression through the common regulatory region containing multiple xenobiotic-responsive elements. Biochem. Pharmacol. 2008, 76, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.; Luo, H.B.; Zheng, Y.; Cheng, Y.K.; Cai, Z. Investigation of the metabolism and reductive activation of carcinogenic aristolochic acid in rats. Drug Metab. Dispos. 2007, 35, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Bock, K.W.; Bock-Hennig, B.S. UDP-glucuronosyltransferases (UGTs): From purification of Ah-receptor-inducible UGT1A6 to coordinate regulation of subsets of CYPs, UGTs, and ABC transporters by nuclear receptors. Drug Metab. Rev. 2010, 42, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Yanagiba, Y.; Ito, Y.; Kamijima, M.; Gonzalez, F.J.; Nakajima, T. Octachlorostyrene induces cytochrome P450, UDP-glucuronosyltransferase, and sulfotransferase via the aryl hydrocarbon receptor and constitutive androstane receptor. Toxicol. Sci. 2009, 111, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-A.; Senthilkumar, R.; Rong, F.; Guo, Q. Cardioprotective potential of baicalein: A short review of in vitro and in vivo studies. Pharm. Anal. Acta 2014, 5, 280–284. [Google Scholar]
- Chen, H.; Gao, Y.; Wu, J.; Chen, Y.; Chen, B.; Hu, J.; Zhou, J. Exploring therapeutic potentials of baicalin and its aglycone baicalein for hematological malignancies. Cancer Lett. 2014, 354, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Cui, H.; Behr, M.; Zhang, L.; Zhang, Q.Y.; Yang, W.; Hinson, J.A.; Ding, X. In vivo mechanisms of tissue-selective drug toxicity: Effects of liver-specific knockout of the NADPH-cytochrome P450 reductase gene on acetaminophen toxicity in kidney, lung, and nasal mucosa. Mol. Pharmacol. 2005, 67, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.; Bresnick, E. Regulation of the constitutive expression of the human CYP1A2 gene: Cis elements and their interactions with proteins. Mol. Pharmacol. 1995, 47, 677–685. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, K.; Feng, C.; Li, C.; Yao, J.; Xie, X.; Gong, L.; Luan, Y.; Xing, G.; Zhu, X.; Qi, X.; et al. Baicalin Protects Mice from Aristolochic Acid I-Induced Kidney Injury by Induction of CYP1A through the Aromatic Hydrocarbon Receptor. Int. J. Mol. Sci. 2015, 16, 16454-16468. https://doi.org/10.3390/ijms160716454
Wang K, Feng C, Li C, Yao J, Xie X, Gong L, Luan Y, Xing G, Zhu X, Qi X, et al. Baicalin Protects Mice from Aristolochic Acid I-Induced Kidney Injury by Induction of CYP1A through the Aromatic Hydrocarbon Receptor. International Journal of Molecular Sciences. 2015; 16(7):16454-16468. https://doi.org/10.3390/ijms160716454
Chicago/Turabian StyleWang, Ke, Chenchen Feng, Chenggang Li, Jun Yao, Xiaofeng Xie, Likun Gong, Yang Luan, Guozhen Xing, Xue Zhu, Xinming Qi, and et al. 2015. "Baicalin Protects Mice from Aristolochic Acid I-Induced Kidney Injury by Induction of CYP1A through the Aromatic Hydrocarbon Receptor" International Journal of Molecular Sciences 16, no. 7: 16454-16468. https://doi.org/10.3390/ijms160716454
APA StyleWang, K., Feng, C., Li, C., Yao, J., Xie, X., Gong, L., Luan, Y., Xing, G., Zhu, X., Qi, X., & Ren, J. (2015). Baicalin Protects Mice from Aristolochic Acid I-Induced Kidney Injury by Induction of CYP1A through the Aromatic Hydrocarbon Receptor. International Journal of Molecular Sciences, 16(7), 16454-16468. https://doi.org/10.3390/ijms160716454