
Double Variational Binding—(SMILES) Conformational Analysis by Docking Mechanisms for Anti-HIV Pyrimidine Ligands


Abstract
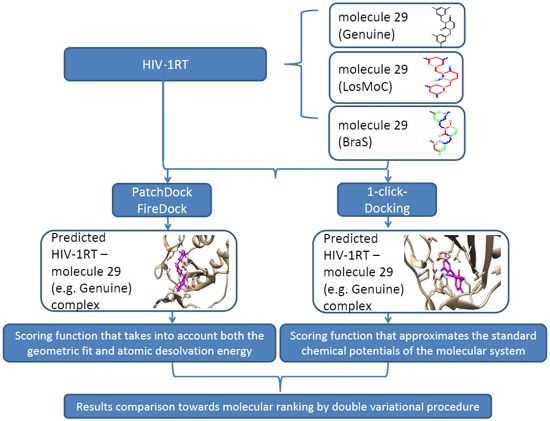
:
1. Introduction
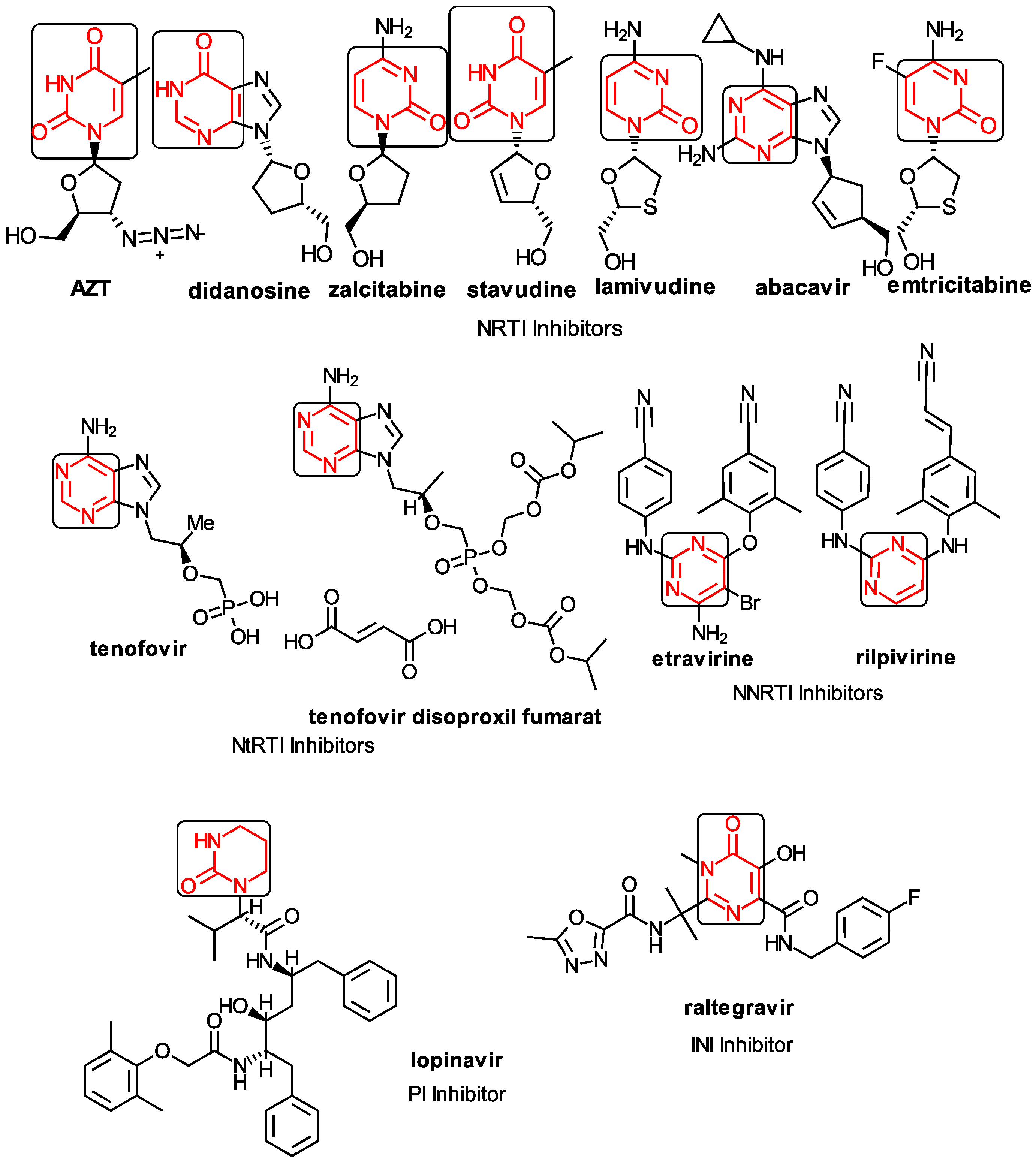
1.1. The General Anti-HIV Context
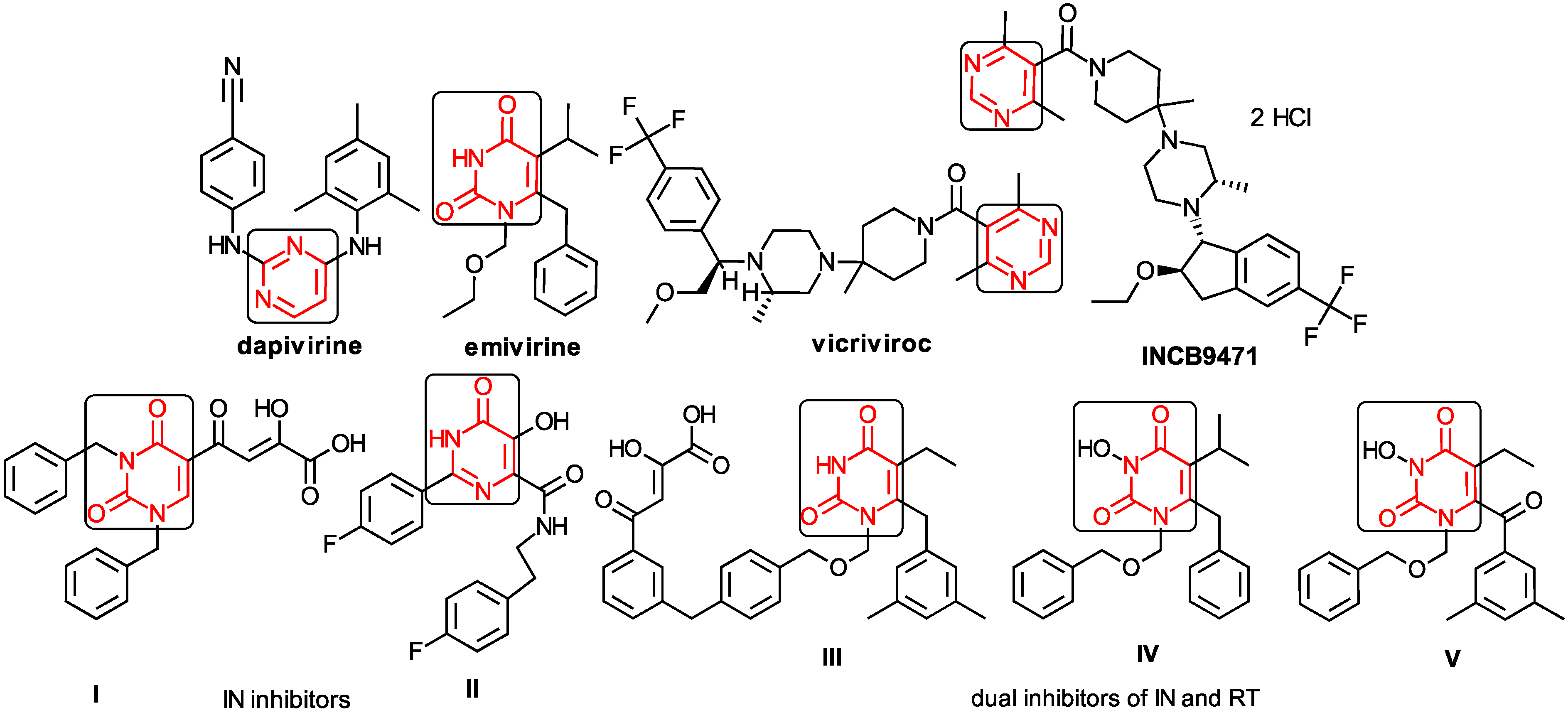
1.2. The Anti-HIV Pyrimidine Derivatives

- ➢
- ➢
- ➢
- ➢

- ➢
- ➢
1.3. The Anti-HIV Mechanisms of Action
- ➢
- The protein/solvent interface is close to Pro236, Val106 and Leu234;
- ➢
- The largely open region in front of Lys101, Lys103, Glu138, and Val179 is considered to form the entrance channel for the NNRTI binding site;
- ➢
- The tunnel is lined by Tyr181, Tyr188, Trp229, and Phe227, which leads towards the polymerase active site;
- ➢
- The groove is lined by Phe227, Tyr318, Pro225, Pro236.
- ➢
- The restriction of thumb mobility;
- ➢
- Distortion of the catalytic triad;
- ➢
- Repositioning of the primer grip;
- ➢
- and loosening the thumb and fingers clamp.
1.4. The NNRTIs–RT Basic Interactions
- ➢
- Hydrophobic sandwiches;
- ➢
- A characteristic hydrogen bond with the Lys101 main-chain carbonyl;
- ➢
- And water-mediated hydrogen bonds.
1.5. Overview of the Present Study
- ➢
- Section 2 presents the working pyrimidine series, their structural roots, as well as their SMILES (simplified molecular input line entry system) conformations, which were created via the controlled breaking of chemical bonds;
- ➢
- The generated longest SMILES molecular chain (LoSMoC) and Branching SMILES (BraS) cases are further considered in Section 3 as the variational transformation into anti-HIV docking action;
- ➢
- The Section 4 interprets the computational docking results in variational Genuine-LoSMoC-Branching (BraS) form while selecting the most versatile pyrimidine molecule able to change its conformation (variationally).
2. SMILES of Anti-HIV Pyrimidines
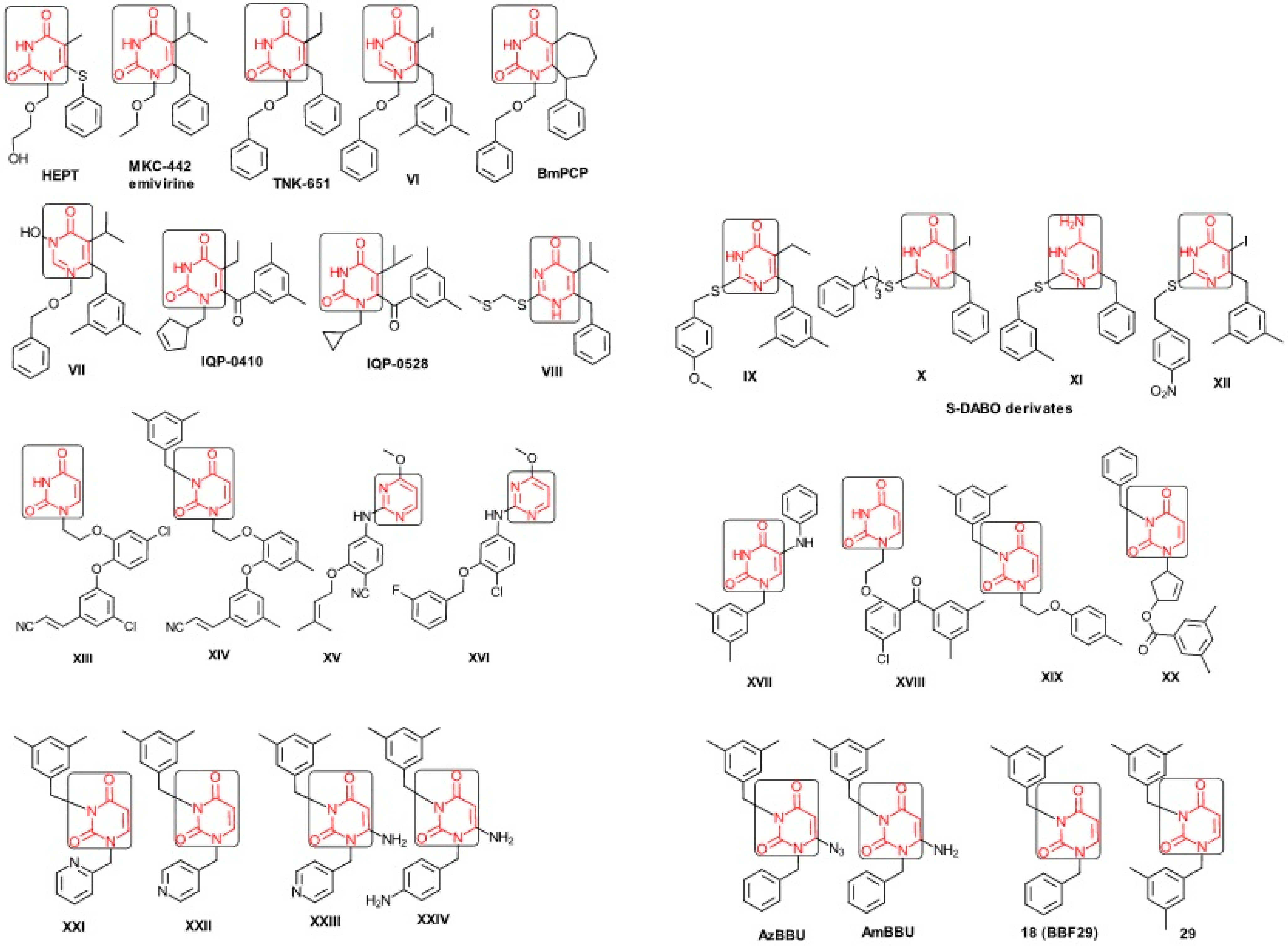
2.1. Presenting the Anti-HIV HEPT Derivatives
- ➢
- The compound VI (1-benzyloxymethyl-6-(3,5-dimethylbenzyl)-5-iodouracil), has potent NNRTI activity against HIV-1 strains resistance (through a halogen at the C-5 position and meta-substituents on the C-6 aromatic moiety) [60];
- ➢
- The compound BmPCP (1-[(benzyloxy)methyl]-9-phenyl-6,7,8,9-tetrahydro-1H-cyclohepta[d]pyrimidine-2,4-(3H,5H)-dione);
- ➢
- The compound VII (6-benzyl-1-(benzyloxymethyl)-3-hydroxy-5-isopropyl-uracil) which proved to be a potential dual inhibitor behaving both as NNRTI and INI alike;
- ➢

- ➢
- The compounds XXI (3-(3,5-dimethylbenzyl)-1-(2-pyridinylmethyl)-2,4(1H,3H)-pyrimidinedione) and XXII (3-(3,5-dimethylbenzyl)-1-(4-pyridinylmethyl)-2,4(1H,3H)-pyrimidinedione);
- ➢
- The foremost representative compounds AzBBU (6-azido-1-benzyl-3-(3,5-dimethylbenzyl) uracil) presumed to be reduced by metabolic pathway in AmBBU (6-amino-1-benzyl-3-(3,5-dimethylbenzyl)uracil;
- ➢
- The derivates of AmBBU as compounds XXIII (6-amino-3-(3,5-dimethylbenzyl)-1-(4-pyridinylmethyl)-uracil and XXIV (6-Amino-3-(3,5-dimethylbenzyl)-1-(4-aminobenzyl)-uracil.
2.2. SMILES Forms for Working HEPT Derivatives

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 principal SMILES chain;
principal SMILES chain;  secondary SMILES branch;
secondary SMILES branch;  tertiary SMILES branch;
tertiary SMILES branch;  quaternary SMILES branch; = double bond; # triple bond; /,\ directional bonds; ( ) branch; C, N, F, S, I–atoms present in the molecule; c, n–atoms place in an aromatic ring; C1/2/3, N1/2, c1/2/3, n2–connectivity points [97,98,99].
quaternary SMILES branch; = double bond; # triple bond; /,\ directional bonds; ( ) branch; C, N, F, S, I–atoms present in the molecule; c, n–atoms place in an aromatic ring; C1/2/3, N1/2, c1/2/3, n2–connectivity points [97,98,99].
| No. | Structure 2D | IUPAC Name | MW | AIDS Code | SMILES Configurations | |
|---|---|---|---|---|---|---|
| LoSMoC | Code LoSMoC | |||||
| BraS | Code BraS | |||||
| 1 |  | [3-(2-Methyl-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile | 255.28 | AIDS352092 |  | N#CCN1/C=C\C(=O) N(C1=O)Cc2ccc(C)c(C)c2 |
 | O=C1N(Cc(c(C)cc2)cc2) C(N(/C=C1\)CC#N)=O | |||||
| 2 |  | [3-(3-Methyl-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile | 255.28 | AIDS352093 |  | N#CCN1/C=C\C(=O) N(C1=O)Cc2cccc(C)c2 |
 | O=C1N(Cc(cc(C)c2)cc2) C(N(/C=C1\)CC#N)=O | |||||
| 3 |  | [3-(4-Methyl-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile | 255.28 | AIDS352094 |  | N#CCN1/C=C\C(=O) N(C1=O)Cc2ccc(C)cc2 |
 | O=C1N(Cc(ccc2C)cc2) C(N(/C=C1\)CC#N)=O | |||||
| 4 |  | [3-(2,4-Dimethyl-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile | 269.30 | AIDS352888 |  | N#CCN1/C=C\C(=O) N(C1=O)Cc2ccc(C)cc2C |
 | O=C1N(Cc2c(cc(cc2)C)C) C(N(/C=C1\)CC#N)=O | |||||
| 5 |  | [3-(2,5-Dimethyl-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile | 269.30 | AIDS352889 |  | N#CCN1/C=C\C(=O) N(C1=O)Cc2cc(C)ccc2C |
 | O=C1N(Cc(cc(C)c2)c(c2)C) C(N(/C=C1\)CC#N)=O | |||||
| 6 |  | [3-(2,6-Dimethyl-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile | 269.30 | AIDS352890 |  | N#CCN1/C=C\C(=O) N(C1=O)Cc2c(C)cccc2C |
 | O=C1N(Cc(c(C)cc2)c(C)c2) C(N(/C=C1\)CC#N)=O | |||||
| 7 |  | [3-(3,5-Dimethyl-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile | 269.30 | AIDS352095 |  | N#CCN1/C=C\C(=O) N(C1=O)Cc2cc(C)cc(C)c2 |
 | O=C1N(Cc(cc(C)c2)cc2C) C(N(/C=C1\)CC#N)=O | |||||
| 8 |  | [3-(3,4-Dimethyl-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile | 269.30 | AIDS352891 |  | N#CCN1/C=C\C(=O) N(C1=O)Cc2ccc(C)c(C)c2 |
 | O=C1N(Cc(cc(c2C)C)cc2) C(N(/C=C1\)CC#N)=O | |||||
| 9 |  | [3-(2,4,6-Trimethyl-benzyl)- 2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile | 283.33 | AIDS352892 |  | N#CCN1/C=C\C(=O) N(C1=O)Cc2c(C)cc(C)cc2C |
 | O=C1N(Cc2c(cc(cc2C)C)C) C(N(/C=C1\)CC#N)=O | |||||
| 10 |  | [3-(3-Cyanophenyl)methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile | 266.26 | AIDS352893 |  | N#CCN1/C=C\C(=O) N(C1=O)Cc2cccc(c2)C#N |
 | O=C1N(Cc(cc(C#N)c2)cc2) C(N(/C=C1\)CC#N)=O | |||||
| 11 |  | [3-(3,5-Dimethoxy-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile | 301.30 | AIDS352897 |  | N#CCN1/C=C\C(=O)N(C1=O) Cc2cc(OC)cc(c2)OC |
 | O=C1N(Cc(cc2OC)cc(OC)c2) C(N(/C=C1\)CC#N)=O | |||||
| 12 |  | [3-(3,4,5-trimethoxy-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile | 331.33 | AIDS352898 |  | N#CCN1/C=C\C(=O)N(C1=O) Cc2cc(OC)c(OC)c(c2)OC |
 | O=C1N(Cc2cc(c(OC)c(OC)c2)OC) C(N(/C=C1\)CC#N)=O | |||||
| 13 |  | (3-Naphthalen-1-ylmethyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-acetonitrile | 291.31 | AIDS352899 |  | N#CCN1/C=C\C(=O)N (C1=O)Cc3c2ccccc2ccc3 |
 | O=C1N(Cc(c(cc3)c(cc3)c2)cc2) C(N(/C=C1\)CC#N)=O | |||||
| 14 |  | (3-Naphthalen-2-ylmethyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-acetonitrile | 291.31 | AIDS352900 |  | N#CCN1/C=C\C(=O)N (C1=O)Cc3cc2ccccc2cc3 |
 | O=C1N(Cc(cc(ccc3)c2c3)cc2) C(N(/C=C1\)CC#N)=O | |||||
| 15 |  | (3-Biphenyl-4-ylmethyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-acetonitrile | 317.35 | AIDS352901 |  | N#CCN1/C=C\C(=O)N (C1=O)Cc2ccc(cc2)c3ccccc3 |
 | O=C1N(Cc(c2)ccc(c(cc3)ccc3)c2) C(N(/C=C1\)CC#N)=O | |||||
| 16 |  | 1-Benzyl-3-phenyl-1H-pyrimidine-2,4-dione | 278.31 | AIDS352902 |  | c1ccccc1CN2/C=C\C(=O) N(C2=O)c3ccccc3 |
 | O=C1N(c(cc2)ccc2)C(N(/C=C1\) Cc(ccc3)cc3)=O | |||||
| 17 |  | 1,3-Dibenzyl-1H-pyrimidine-2,4-dione | 292.34 | AIDS352903 |  | c1ccccc1CN2/C=C\C(=O) N(C2=O)Cc3ccccc3 |
 | O=C1N(Cc(ccc2)cc2)C (N(/C=C1\)Cc(ccc3)cc3)=O | |||||
| 18 |  | 1-Benzyl-3-(3,5-dimethyl-benzyl)-1H-pyrimidine-2,4-dione | 320.39 | AIDS352096 |  | c1ccccc1CN2/C=C\C(=O) N(C2=O)Cc3cc(C)cc(C)c3 |
 | O=C1N(Cc(cc(C)c2)cc2C)C (N(/C=C1\)Cc(ccc3)cc3)=O | |||||
| 19 |  | 1-Benzyl-3-(4,6-dimethyl-pyridin-2-ylmethyl)-1H-pyrimidine-2,4-dione | 321.38 | AIDS352904 |  | c1ccccc1CN2/C=C\C(=O) N(C2=O)Cc3nc(C)cc(C)c3 |
 | O=C1N(Cc(cc(C)c2)nc2C)C (N(/C=C1\)Cc(ccc3)cc3)=O | |||||
| 20 |  | 1-Benzyl-3-(3,5-dimethyl-benzyl)-5-methyl-1H-pyrimidine-2,4-dione | 334.42 | AIDS352905 |  | c1ccccc1CN2/C=C\(C)C(=O) N(C2=O)Cc3cc(C)cc(C)c3 |
 | c1ccccc1CN2/C=C\(C)C(=O) N(C2=O)Cc3cc(C)cc(C)c3 | |||||
| 21 |  | 1-Benzyl-3-(3,5-dimethyl-benzyl)-5-iodo-1H-pyrimidine-2,4-dione | 446.29 | AIDS352906 |  | c1ccccc1CN2/C=C\(I)C(=O) N(C2=O)Cc3cc(C)cc(C)c3 |
 | O=C1N(Cc(cc(C)c2)cc2C) C(N(/C=C1\I)Cc(ccc3)cc3)=O | |||||
| 22 |  | 1-(2,6-Difluoro-benzyl)-3-phenyl-1H-pyrimidine-2,4-dione | 314.29 | AIDS352907 |  | Fc1cccc(F)c1CN2/C=C\C (=O)N(C2=O)c3ccccc3 |
 | O=C1N(c(cc2)ccc2)C(N(/C=C1\) Cc(c(F)cc3)c(F)c3)=O | |||||
| 23 |  | 1-(2,6-Difluoro-benzyl)-3-(3,5-dimethyl-benzyl)-1H-pyrimidine-2,4-dione | 356.37 | AIDS352908 |  | Fc1cccc(F)c1CN2/C=C\C(=O) N(C2=O)Cc3cc(C)cc(C)c3 |
 | O=C1N(Cc(cc(C)c2)cc2C)C (N(/C=C1\)Cc(c(F)cc3)c(F)c3)=O | |||||
| 24 |  | 1-(2,6-Difluoro-benzyl)-3-(4,6-dimethyl-pyridin-2-ylmethyl)-1H-pyrimidine-2,4-dione | 357.36 | AIDS352909 |  | Fc1cccc(F)c1CN2/C=C\C(=O) N(C2=O)Cc3nc(C)cc(C)c3 |
 | O=C1N(Cc(cc(C)c2)nc2C)C (N(/C=C1\)Cc(c(F)cc3)c(F)c3)=O | |||||
| 25 |  | 1-(2,6-Difluoro-benzyl)-3-(2,6-dimethyl-pyridin-4-ylmethyl)-1H-pyrimidine-2,4-dione | 357.36 | AIDS352910 |  | Fc1cccc(F)c1CN2/C=C\C(=O) N(C2=O)Cc3cc(C)nc(C)c3 |
 | O=C1N(Cc(cc(C)n2)cc2C)C(N(/C=C1\) Cc(c(F)cc3)c(F)c3)=O | |||||
| 26 |  | 1,3-Bis-(2,6-difluoro-benzyl)-1H-pyrimidine-2,4-dione | 364.30 | AIDS352911 |  | Fc1cccc(F)c1CN2/C=C\C(=O) N(C2=O)Cc3c(F)cccc3F |
 | O=C1N(Cc(c(F)cc2)c(F)c2)C (N(/C=C1\)Cc(c(F)cc3)c(F)c3)=O | |||||
| 27 |  | 3-(3,5-Dimethyl-benzyl)-1-phenethyl-1H-pyrimidine-2,4-dione | 334.42 | AIDS352912 |  | c1ccccc1CCN2/C=C\C(=O) N(C2=O)Cc3cc(C)cc(C)c3 |
 | O=C1N(Cc(cc(C)c2)cc2C) C(N(/C=C1\)CCc(cccc3)c3)=O | |||||
| 28 |  | 3-(3,5-Dimethyl-benzyl)-1-prop-2-ynyl-1H-pyrimidine-2,4-dione | 268.32 | AIDS352913 |  | C#CCN1/C=C\C(=O) N(C1=O)Cc2cc(C)cc(C)c2 |
 | C#CCN1/C=C\C(=O) N(C1=O)Cc2cc(C)cc(C)c2 | |||||
| 29 |  | 1,3-Bis-(3,5-dimethyl-benzyl)-1H-pyrimidine-2,4-dione | 348.44 | AIDS352914 |  | c1c(C)cc(C)cc1CN2/C=C\C(=O) N(C2=O)Cc3cc(C)cc(C)c3 |
 | O=C1N(Cc(cc(C)c2)cc2C)C (N(/C=C1\)Cc(cc(cc3C)C)c3)=O | |||||
| 30 |  | [3-(3,5-Dimethyl-benzyl)-2-oxo-4-thioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile | 285.36 | AIDS352915 |  | N#CCN1/C=C\C(=S)N (C1=O)Cc2cc(C)cc(C)c2 |
 | S=C1N(Cc(cc(C)c2)cc2C) C(N(/C=C1\)CC#N)=O | |||||
| 31 |  | 1-Benzyl-3-(3,5-dimethyl-benzyl)-4-thioxo-3,4-dihydro-1H-pyrimidin-2-one | 336.45 | AIDS352916 |  | c1ccccc1CN2/C=C\C(=S) N(C2=O)Cc3cc(C)cc(C)c3 |
 | S=C1N(Cc(cc(C)c2)cc2C) C(N(/C=C1\)Cc(ccc3)cc3)=O | |||||
| 32 |  | 1-(2,6-Difluoro-benzyl)-3-(3,5-dimethyl-benzyl)-4-thioxo-3,4-dihydro-1H-pyrimidin-2-one | 372.43 | AIDS352917 |  | Fc1cccc(F)c1CN2/C=C\C(=S) N(C2=O)Cc3cc(C)cc(C)c3 |
 | S=C1N(Cc(cc(C)c2)cc2C) C(N(/C=C1\)Cc(c(F)cc3)c(F)c3)=O | |||||
- The longest SMILES molecular chain (LoSMoC) is assumed to be the first stage in intermediary molecular defolding targeting the receptor. It is obtained by breaking one bond in every aromatic ring in the original molecule. The resulting “molecule” is displayed as a sort of 2D form for the original molecule, so casting a kind of “fractalic” chain; the maximum SMILES chain in LoSMoC is presumably responsible for best transport/transduction of ligand molecules through cellular (lipidic) walls. Afterwards they may be released with a modified structure due to their further ionization upon the interaction with cellular layers. Accordingly, another form of SMILES is generated and next to be considered, as follows;
- The Branching SMILES (BraS) represents the second conformation-phase of molecular defolding. It is obtained by ligand bonds’ breaking such that many “bays” are formed, yet with consistent “arms” linking the short molecular “skeleton” aiming to favor the binding with receptor in its pockets. Accordingly, the branching is not necessary in the same points of molecules through a series, yet an ”equilibrium” between maximum branching and stericity of branches accounts for the final BraS. For instance, a long branch adjacent to a short one will make an “anchor” not strong enough to bind the receptor pocket. Therefore, the “branching principle” requires having the equilibrated anchor-clefs by themselves. As such, the branching up to fourth order is performed for molecules in Table 1.
- ➢
- By the computational proof of the uncatalyzed racemization process where the openings and closures of the pyrimidinic nuclei happen just there where the above LoSMoc take places (Table 1). Therefore the concerned binding breaks go through a sort of SMILES transformations with a lower energy, following the principle of favoring the longest chain in molecular configuration. This is already a sort of structural variational principle in chemical bonding [108];
- ➢
- By the recent QSARINS-Chem model for QSAR studies recognizing the role of SMILES canonical rules in correctly assessing the query and parsing the structure-activity algorithm (for the LoSMoC and BraS conformations, for instance) [109].
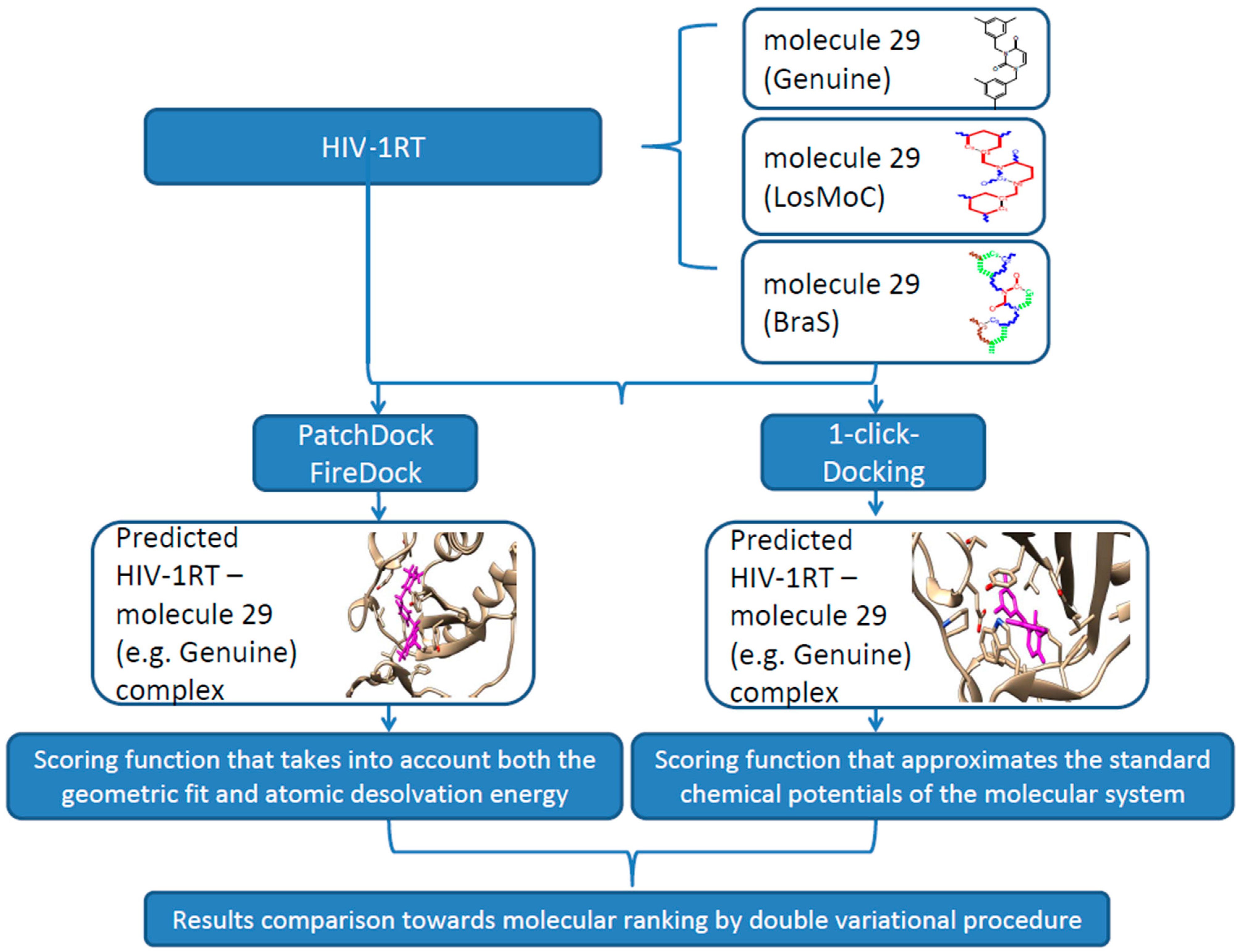
3. Docking of Anti-HIV 1,3-Disubstituted Uracil Derivatives
3.1. Docking Algorithm
3.2. Docking Results

| Molecule Genuine, No. | Molecular Area (Å2) | Molecular Volume (Å3) | PatchDock | 1-Click Docking | ||
|---|---|---|---|---|---|---|
| Global Energy (kcal) | Interface Area (Å2) | Binding Affinity (kcal/mol) | Toxicity | |||
| 1 | 318.3 | 358.2 | −20.44 | 442.80 | −9.30 | Potentially toxic |
| 2 | 333.7 | 386.2 | −22.24 | 511.40 | −9.40 | Potentially toxic |
| 3 | 326.8 | 363.1 | −21.83 | 450.40 | −8.30 | Potentially toxic |
| 4 | 342.4 | 388.7 | −23.85 | 489.70 | −8.70 | Potentially toxic |
| 5 | 342.4 | 390.2 | −25.24 | 481.00 | −9.40 | Potentially toxic |
| 6 | 331.6 | 387.0 | −22.86 | 498.10 | −9.60 | Potentially toxic |
| 7 | 351.9 | 396.1 | −22.67 | 505.00 | −9.70 | Potentially toxic |
| 8 | 345.3 | 391.0 | −22.97 | 505.10 | −9.10 | Potentially toxic |
| 9 | 356.1 | 419.9 | −24.63 | 546.00 | −9.20 | Potentially toxic |
| 10 | 318.2 | 345.7 | −20.48 | 467.00 | −5.80 | Potentially toxic |
| 11 | 369.7 | 417.4 | −23.89 | 549.00 | −8.20 | Potentially toxic |
| 12 | 398.3 | 462.5 | −27.82 | 567.70 | −7.40 | Potentially toxic |
| 13 | 351.1 | 395.9 | −25.03 | 526.70 | −7.20 | Potentially toxic |
| 14 | 353.0 | 393.1 | −23.19 | 503.40 | −6.50 | Potentially toxic |
| 15 | 392.1 | 438.1 | −24.34 | 560.90 | −7.70 | Potentially toxic |
| 16 | 349.9 | 393.7 | −23.11 | 511.20 | −10.4 | Nontoxic |
| 17 | 371.5 | 422.9 | −25.89 | 546.00 | −10.2 | Nontoxic |
| 18 | 422.3 | 489.4 | −27.34 | 587.30 | −11.3 | Nontoxic |
| 19 | 414.9 | 483.3 | −26.99 | 587.80 | −10.8 | Nontoxic |
| 20 | 444.0 | 515.1 | −28.83 | 675.00 | −10.7 | Nontoxic |
| 21 | 433.1 | 501.9 | −26.78 | 648.00 | −11.8 | Nontoxic |
| 22 | 354.3 | 399.4 | −24.97 | 509.10 | −10.00 | Nontoxic |
| 23 | 426.6 | 496.1 | −23.31 | 561.70 | −11.10 | Nontoxic |
| 24 | 424.4 | 488.1 | −28.24 | 616.30 | −8.00 | Nontoxic |
| 25 | 422.1 | 486.2 | −24.82 | 616.30 | −10.50 | Nontoxic |
| 26 | 380.9 | 438.0 | −23.53 | 543.50 | −10.40 | Nontoxic |
| 27 | 439.4 | 531.0 | −26.85 | 657.80 | −11.10 | Nontoxic |
| 28 | 363.9 | 406.1 | −25.77 | 512.00 | −9.50 | Potentially toxic |
| 29 | 427.9 | 556.1 | −29.82 | 707.10 | −11.50 | Potentially toxic |
| 30 | 354.4 | 399.9 | −23.58 | 506.00 | −9.20 | Potentially toxic |
| 31 | 427.7 | 493.1 | −26.48 | 594.70 | −11.00 | Potentially toxic |
| 32 | 427.7 | 497.8 | −25.84 | 630.80 | −11.3 | Potentially toxic |
| Molecule Branched | Molecular Area (Å2) | Molecular Volume (Å3) | PatchDock | 1-Click Docking | ||
|---|---|---|---|---|---|---|
| Global Energy (kcal) | Interface Area (Å2) | Binding Affinity (kcal/mol) | Toxicity | |||
| 1 | 331.5 | 380.4 | −22.09 | 473.00 | −7.40 | Potentially toxic |
| 2 | 336.3 | 376.7 | −20.69 | 488.80 | −4.60 | Potentially toxic |
| 3 | 335.9 | 375.7 | −23.66 | 492.60 | −4.70 | Potentially toxic |
| 4 | 347.2 | 404.5 | −25.32 | 519.20 | −6.00 | Potentially toxic |
| 5 | 349.7 | 397.6 | −23.10 | 505.20 | −5.80 | Potentially toxic |
| 6 | 346.1 | 400.4 | −22.69 | 489.00 | −7.60 | Potentially toxic |
| 7 | 361.3 | 410.0 | −24.06 | 534.60 | −7.40 | Potentially toxic |
| 8 | 356.3 | 406.3 | −24.02 | 509.01 | −7.60 | Potentially toxic |
| 9 | 365.2 | 431.1 | −26.85 | 536.70 | −8.10 | Potentially toxic |
| 10 | 327.4 | 359.4 | −21.72 | 454.00 | −7.00 | Potentially toxic |
| 11 | 380.0 | 424.9 | −22.46 | 529.70 | −5.20 | Potentially toxic |
| 12 | 406.1 | 474.6 | −24.31 | 591.40 | −5.50 | Potentially toxic |
| 13 | 384.7 | 409.6 | −22.06 | 506.80 | −7.90 | Potentially toxic |
| 14 | 387.3 | 413.5 | −22.16 | 510.20 | – | Potentially toxic |
| 15 | 400.3 | 446.4 | −28.20 | 605.70 | – | Potentially toxic |
| 16 | 370.1 | 412.6 | −22.53 | 502.10 | −8.20 | Potentially toxic |
| 17 | 397.1 | 449.0 | −27.21 | 580.10 | −8.30 | Potentially toxic |
| 18 | 442.5 | 509.4 | −26.17 | 655.10 | −6.10 | Potentially toxic |
| 19 | 440.3 | 501.3 | −28.46 | 633.20 | −8.00 | Potentially toxic |
| 20 | 462.3 | 539.0 | −26.17 | 644.20 | −8.60 | Potentially toxic |
| 21 | 448.7 | 515.7 | −23.04 | 620.80 | −6.30 | Potentially toxic |
| 22 | 372.3 | 414.4 | −23.59 | 515.10 | −8.00 | Potentially toxic |
| 23 | 440.4 | 509.4 | −21.71 | 640.60 | −8.30 | Potentially toxic |
| 24 | 438.2 | 501.1 | −24.35 | 635.40 | −8.20 | Potentially toxic |
| 25 | 439.6 | 501.1 | −24.44 | 647.80 | −7.80 | Potentially toxic |
| 26 | 397.7 | 451.8 | −23.62 | 561.50 | −8.00 | Potentially toxic |
| 27 | 461.2 | 536.0 | −26.39 | 538.50 | −8.20 | Potentially toxic |
| 28 | 368.2 | 413.0 | −22.06 | 522.40 | −7.70 | Potentially toxic |
| 29 | 486.9 | 569.1 | −25.86 | 582.70 | −8.20 | Potentially toxic |
| 30 | 362.7 | 410.5 | −24.05 | 544.80 | −7.80 | Potentially toxic |
| 31 | 442.9 | 512.5 | −25.96 | 638.20 | −8.20 | Potentially toxic |
| 32 | 445.6 | 515.6 | −27.96 | 648.00 | −8.30 | Potentially toxic |
| Molecule Branched | Molecular Area (Å2) | Molecular Volume (Å3) | PatchDock | 1-Click Docking (AutoDock Vina) | ||
|---|---|---|---|---|---|---|
| Global Energy (kcal) | Interface Area (Å2) | Binding Affinity (kcal/mol) | Toxicity | |||
| 1 | 326.5 | 369.3 | −21.18 | 485.20 | −7.60 | Potentially toxic |
| 2 | 336.5 | 376.9 | −23.16 | 493.90 | −8.50 | Potentially toxic |
| 3 | 336.0 | 375.8 | −25.23 | 460.00 | −4.80 | Potentially toxic |
| 4 | 347.9 | 404.4 | −24.99 | 501.60 | −7.90 | Potentially toxic |
| 5 | 349.7 | 397.6 | −23.10 | 505.20 | −8.00 | Potentially toxic |
| 6 | 346.3 | 400.3 | −23.43 | 514.70 | −7.80 | Potentially toxic |
| 7 | 361.2 | 409.9 | −23.06 | 547.80 | −8.50 | Potentially toxic |
| 8 | 356.2 | 406.2 | −24.23 | 534.00 | −7.50 | Potentially toxic |
| 9 | 365.2 | 431.7 | −27.04 | 549.00 | −8.20 | Potentially toxic |
| 10 | 327.4 | 359.5 | −23.38 | 631.10 | −7.40 | Potentially toxic |
| 11 | 380.0 | 424.9 | −22.46 | 529.70 | −4.40 | Potentially toxic |
| 12 | 406.6 | 474.7 | −26.03 | 582.90 | −5.40 | Potentially toxic |
| 13 | 384.8 | 409.6 | −21.70 | 478.10 | – | Potentially toxic |
| 14 | 387.3 | 413.4 | −21.12 | 505.00 | −8.10 | Potentially toxic |
| 15 | 400.4 | 446.5 | −26.23 | 551.60 | −5.90 | Potentially toxic |
| 16 | 370.7 | 413.1 | −23.37 | 529.00 | −4.30 | Potentially toxic |
| 17 | 397.5 | 451.6 | −24.88 | 559.50 | – | Potentially toxic |
| 18 | 442.5 | 509.1 | −27.12 | 669.40 | −5.10 | Potentially toxic |
| 19 | 440.7 | 501.7 | −27.25 | 611.10 | −4.90 | Potentially toxic |
| 20 | 462.2 | 538.5 | −26.01 | 611.90 | −4.60 | Potentially toxic |
| 21 | 448.7 | 517.7 | −21.27 | 652.50 | −5.60 | Potentially toxic |
| 22 | 372.5 | 414.5 | −23.57 | 533.30 | −8.60 | Potentially toxic |
| 23 | 440.5 | 509.2 | −23.94 | 592.30 | −5.10 | Potentially toxic |
| 24 | 438.0 | 500.4 | −23.90 | 626.80 | −5.00 | Potentially toxic |
| 25 | 439.3 | 501.2 | −23.70 | 596.90 | −9.30 | Potentially toxic |
| 26 | 399.5 | 454.1 | −24.27 | 576.00 | −10.10 | Potentially toxic |
| 27 | 461.4 | 536.5 | −27.21 | 677.00 | −5.60 | Potentially toxic |
| 28 | 368.4 | 412.9 | −22.02 | 540.60 | −8.20 | Potentially toxic |
| 29 | 486.7 | 568.8 | −22.23 | 681.00 | −8.80 | Potentially toxic |
| 30 | 362.9 | 410.4 | −24.80 | 518.90 | −4.20 | Potentially toxic |
| 31 | 443.0 | 512.8 | −25.04 | 655.50 | −5.00 | Potentially toxic |
| 32 | 445.7 | 515.7 | −23.38 | 631.10 | – | Potentially toxic |
4. Discussion: Variational Binding-Conformational Analysis
4.1. The Double Variational Output
- ➢
- Minimum (in negative, so favoring the binding) energy (either as affinity and/or global) associated with a toxically potent molecule highly recommends that structure for the binding purpose, according with the performed docking algorithm.
- ➢
- Binding variational procedure across the various conformations of a compound, such as Genuine, LoSMoC and BraS, towards further providing binding-conformational best regarded molecule(s) for the aimed anti-viral activity.
- ➢
- Variational binding affinity procedure selects the following toxically-potent molecules:
- Genuine: 18, 21, 29;
- LoSMoC: 16, 17, 20, 23, 24, 27, 29, 31, 32;
- BraS: 25, 26, 29;
- ➢
- Variational global energy procedure selects the following toxically-potent molecules:
- Genuine: 20, 24, 29;
- LoSMoC: 15, 17, 19, 27, 32;
- BraS: 18, 19, 27;
- ➢
- Now we are in position to identify the “first intersection” regarding the recorded double outputs per configuration (Genuine, LoSMoC, and BraS) while passing from binding affinity to global energy minimums:
- Genuine: 29;
- LoSMoC: 17, 27, 32;
- ➢
- Performing the “second intersection” regarding the multiple outputs inter-configurations (among Genuine, LoSMoC, and BraS) while maintaining either binding affinity or global energy framework:
- 18: Genuine & BraS;
- 19, 27: LoSMoC & BraS;
- 20, 24: Genuine & LoSMoC;
- 29: Genuine, BraS, LoSMoC;
- 29: Genuine, LoSMoC, & BraS;
- 18: Genuine & BraS;
- 17: LoSMoC;
- 19: LoSMoC & BraS;
- 20: Genuine & LoSMoC;
- 24: Genuine & LoSMoC;
- 27: LoSMoC & BraS;
- 32: LoSMoC;



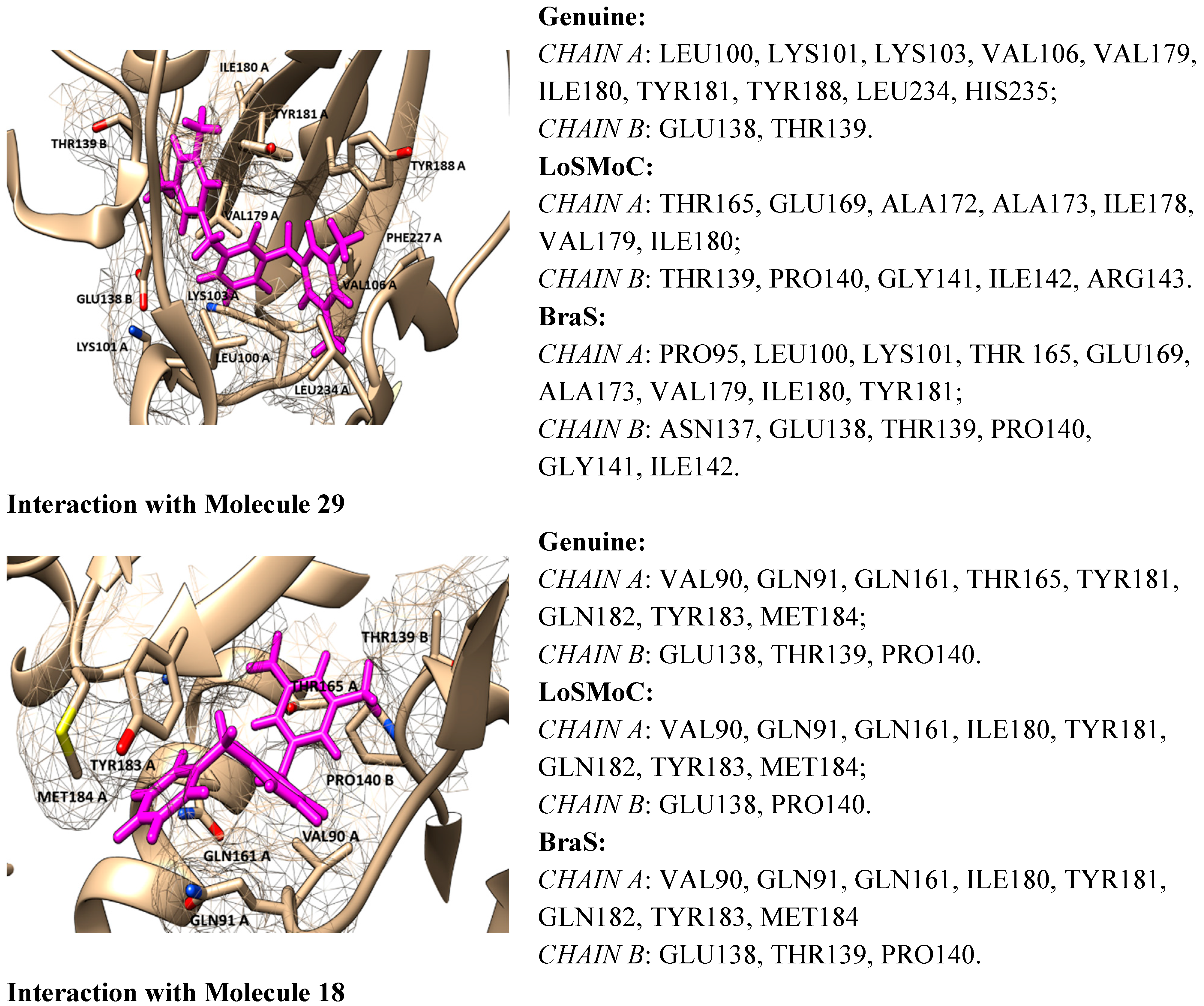
4.2. Discussing the Interaction with Amino Acids
- ➢
- The position of the molecule is in the hydrophobic region of the NNRTI binding site/hydrophobic interactions (by π–π, π-CH, van der Waals contacts) having as two major substituents of the pyrimidine core the residues Tyr181, Tyr188, Phe227, Trp229, His235, Pro238 and/or Val106;
- ➢
- The –CH2– linker of benzyl group or methyl group bound to the benzene ring is positioned closely to Glu138 from the p51 domain of RT, while the pyrimidine core is positioned in the area between Leu100 and Val179;
- ➢
- The formation of one or more H-bonds with Lys101 (and/or Lys103) where there are possible;
- ➢
- The Ar-H interactions with Leu234 are often observed.
- ➢
- For compound 20/LoSMoC: Leu100, Lys101, Lys103, Val106, Val179, Tyr181, Phe227, Trp229, Leu234;
- ➢
- For compound 20/Genuine: Lys101, Lys103, Val106, Val179, Tyr181, Tyr188, Phe227, Trp229, Leu234, His235, Pro236;
- ➢
- For compound 24/LoSMoC: Lys101, Lys103, Val179, Tyr181, Tyr188, Phe227, Glu138B;
- ➢
- For compound 24/Genuine: Leu100, Lys101, Lys103, Val106, Val179, Tyr181, Tyr188, Phe227, Trp229, Leu234, Glu138B.
- ➢
- Leu100 (H) with the central aromatic ring (2-pyrimidine) of AmBBU;
- ➢
- Val106 (H) with 1-benzyl of AmBBU;
- ➢
- Tyr181 (arene) with hydrogen (3-methyl) at 3-(3,5-dimethylbenzyl) of AmBBU;
- ➢
- Trp229 (arene) with 4ʹ-hydrogen of 3-(3,5-dimethylbenzyl) of AmBBU.
- ➢
- The 6-amino group forms a H-bond with Lys 101 (due to water solubility of the 4-aminobenzyl group of XXIV);
- ➢
- The 3,5-dimethylbenzyl moiety enhanced the π–π stacking of the benzene rings of the Tyr181 and Tyr188 residues;
- ➢
- The CH–π interactions are manifested between the methyl group of the 3,5-dimethylbenzyl moiety and Trp229 residue, or between the benzene rings of the 3,5-dimethylbenzyl moiety and Leu234 residue.
- ➢
- LoSMoC favoring cellular penetration;
- ➢
- Branch favoring “binding” to the active site;
- ➢
- and Genuine towards restoring the original molecule, the actually inhibition.
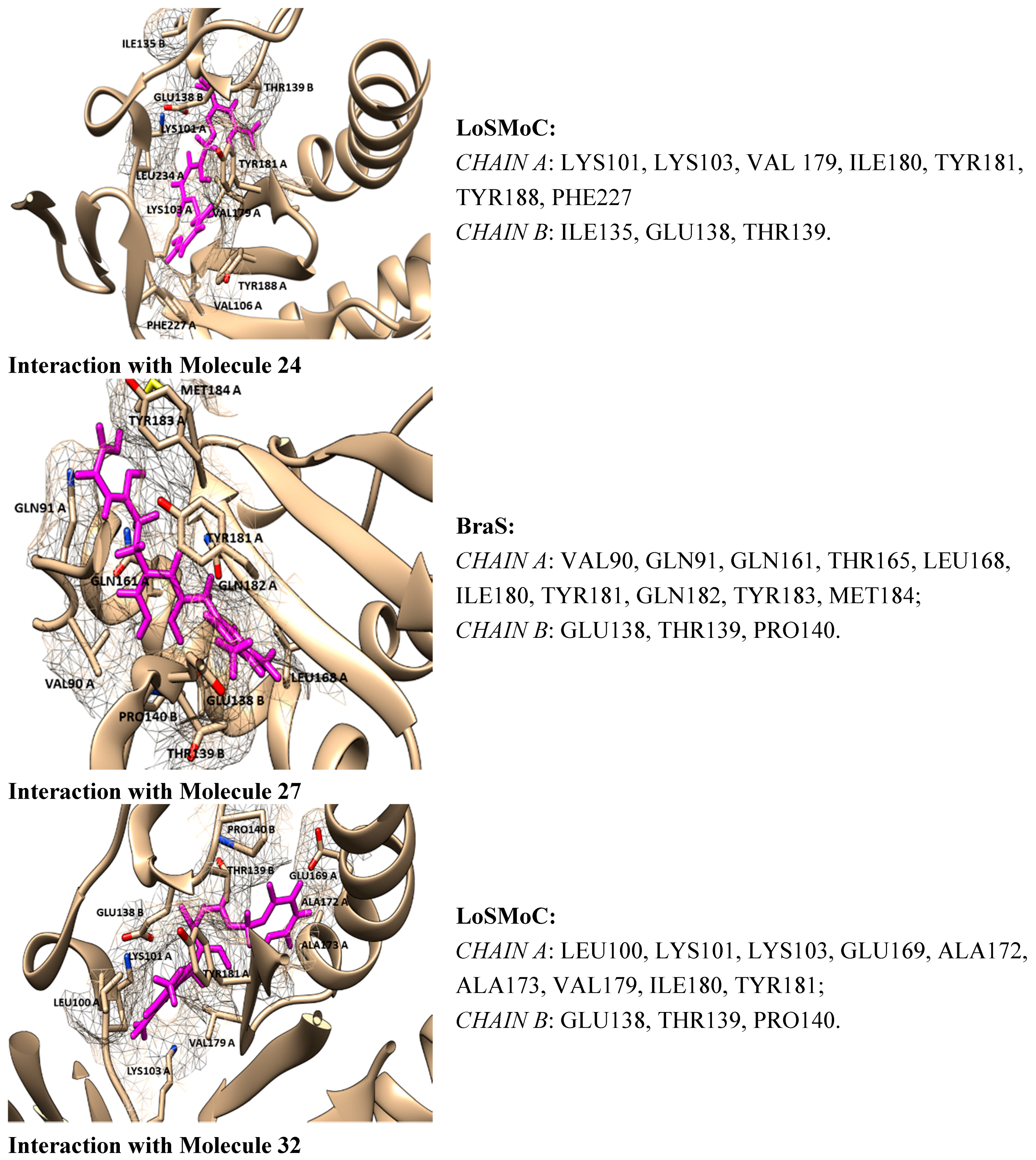
- ➢
- By the LoSMoC configuration it features the specific interactions of pyrimidine NNRTI derivatives only with Val179, so predicting the future position of the pyrimidine core;
- ➢
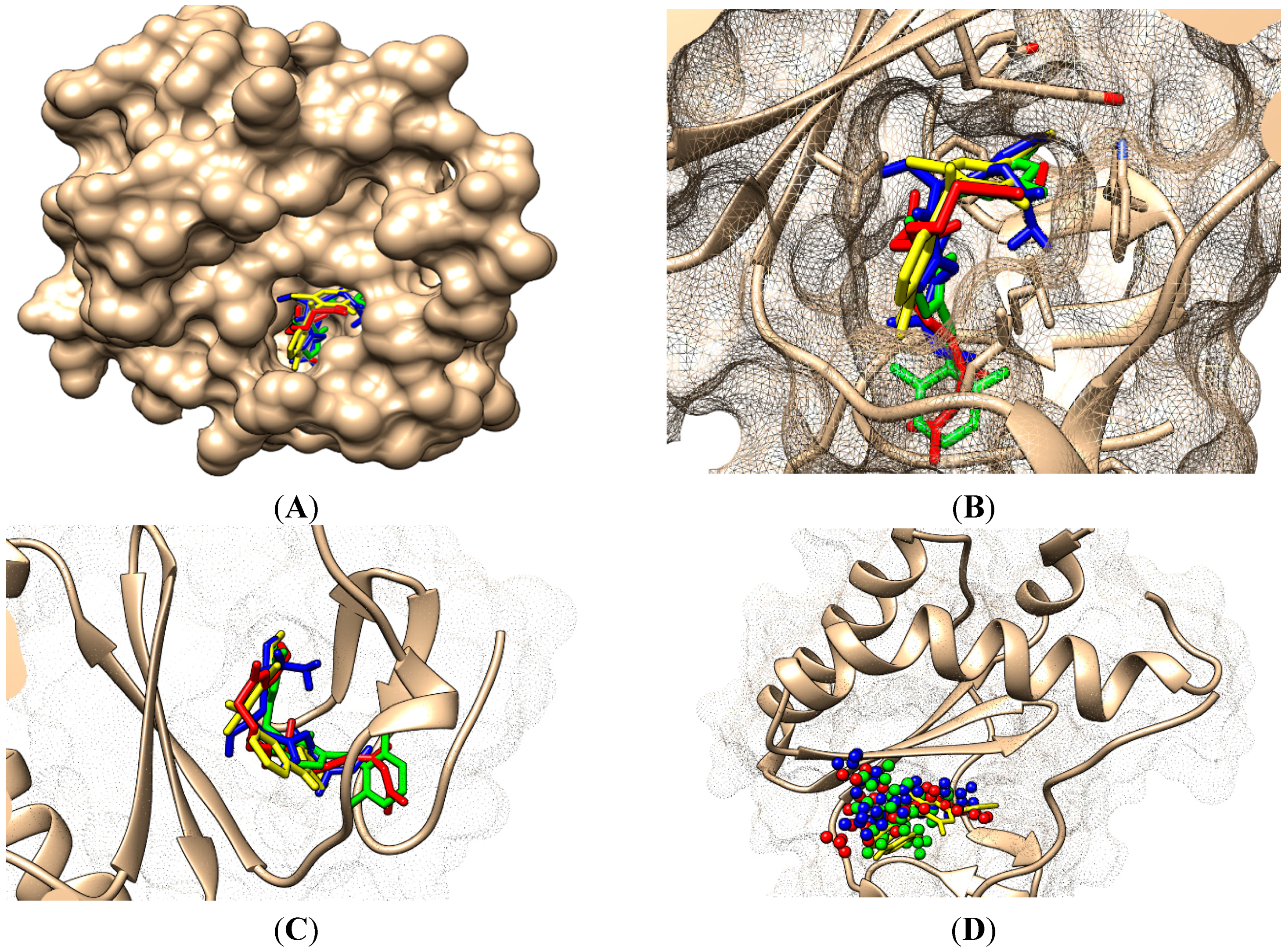
- On its Branch form the presence of new amino acid residues specific to NNRTI-pyrimidines are observed: Glu138B, Lys101, Val179 and Leu100; the latter two are forming the future space where pyrimidine core will be set in, with the specific placement of pyrimidine substituent in the hydrophobic area being delimited, among others in its entry of Figure 5, by Tyr181;
- ➢
- In the Genuine conformation the compound 29 has the correct “U” shape of molecule with a little twist, so keeping in its proximity the same amino acid residues as in the previous Branch form, along the additional ones: Lys103, Ile180, Val106, Tyr188, Leu234, and His235.
- ➢
- Restoring the pyrimidine core in the area between Val179 and Leu100;
- ➢
- Having the –CH2– linker of the benzyl group placed closely to Glu138B;
- ➢
- Having the methyl groups from 3,5-dimethyl-benzyl and the benzene ring deep positioned in the hydrophobic pocket, closed to Val106, Tyr188, Leu234, and His235 (forming hydrophobic interactions with them);
- ➢
- Having the other 3,5-dimethyl-benzyl substituent placed close to Tyr 181 and Ile180.
5. Conclusions

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Halford, B. Aiming for HIV’s weak spot. Chem. Eng. News 2014, 9, 14–21. [Google Scholar]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [PubMed]
- Zhao, Q.; Ma, L.; Jiang, S.; Lu, H.; Liu, S.; He, Y.; Strick, N.; Neamati, N.; Debnath, A.K. Identification of N-phenyl-N′-(2, 2, 6, 6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology 2005, 339, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Schön, A.; Madani, N.; Klein, J.C.; Hubicki, A.; Ng, D.; Yang, X.; Smith, A.B., III; Sodroski, J.; Freire, E. Thermodynamics of binding of a low-molecular-weight CD4 mimetic to HIV-1 gp120. Biochemistry 2006, 45, 10973–10980. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Schön, A.; Freire, E. Optimization of CD4/gp120 inhibitors by thermodynamic-guided alanine-scanning mutagenesis. Chem. Biol. Drug Des. 2013, 81, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Courter, J.R.; Madani, N.; Sodroski, J.; Schön, A.; Freire, E.; Kwong, P.D.; Hendrickson, W.A.; Chaiken, I.M.; LaLonde, J.M.; Smith, A.B., III. Structure-based design, synthesis and validation of CD4-mimetic small molecule inhibitors of HIV-1 entry: Conversion of a viral entry agonist to an antagonist. Acc. Chem. Res. 2014, 47, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Madani, N.; Princiotto, A.M.; Schön, A.; LaLonde, J.; Feng, Y.; Freire, E.; Park, J.; Courter, J.R.; Jones, D.M.; Robinson, J.; et al. CD4-mimetic small molecules sensitize human immunodeficiency virus to vaccine-elicited antibodies. J. Virol. 2014, 88, 6542–6555. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Chien, C.; Timmins, P.; Dennis, A.; Doll, W.; Sandefer, E.; Page, R.; Nettles, R.E.; Zhu, L.; Grasela, D. Compartmental absorption modeling and site of absorption studies to determine feasibility of an extended-release formulation of an HIV-1 attachment inhibitor phosphate ester prodrug. J. Pharm. Sci. 2013, 102, 1742–1751. [Google Scholar] [CrossRef] [PubMed]
- Dudaş, N.A.; Putz, M.V. Pyrimidine derivates with biological activity in anti-HIV therapy. The SPECTRAL-DIAGONAL-SAR approach. Int. J. Chem. Model. 2014, 6, 95–114. [Google Scholar]
- De Clercq, E. The nucleoside reverse transcriptase inhibitors, nonnucleoside reverse transcriptase inhibitors, and protease inhibitors in the treatment of HIV infections (AIDS). Adv. Pharmacol. 2012, 67, 317–358. [Google Scholar]
- Sofia, M.J. Chapter fifteen—Nucleosides and Nucleotides for the Treatment of Viral Diseases. In Annual Reports in Medicinal Chemistry; Desai, M.C., Ed.; Elsevier Inc.: London, UK, 2014; Volume 49, pp. 222–228. [Google Scholar]
- Sahlberg, C.; Zhou, X.X. Development of non-nucleoside reverse transcriptase inhibitors for anti-HIV therapy. Anti-Infect. Agents Med. Chem. 2008, 7, 101–117. [Google Scholar] [CrossRef]
- Esposito, F.; Corona, A.; Tramontano, E. HIV-1 reverse transcriptase still remains a new drug target: Structure, function, classical inhibitors, and new inhibitors with innovative mechanisms of actions. Mol. Biol. Int. 2012, 2012, 586401. [Google Scholar] [CrossRef] [PubMed]
- Usach, I.; Melis, V.; Peris, J.E. Non-nucleoside reverse transcriptase inhibitors: A review on pharmacokinetics, pharmacodynamics, safety and tolerability. J. Int. AIDS Soc. 2013, 16, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ma, L.; Wang, X.; Liu, J. New efficient and flexible synthetic route to emivirine and its analogs. J. Heterocycl. Chem. 2013, 50, 164–168. [Google Scholar] [CrossRef]
- Friend, D.R.; Kiser, P.F. Assessment of topical microbicides to prevent HIV-1 transmission: Concepts, testing, lessons learned. Antivir. Res. 2013, 99, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Chi, G.; Ptak, R.; Neamati, N. HIV integrase inhibitors with nucleobase scaffolds: Discovery of a highly potent anti-HIV agent. J. Med. Chem. 2006, 49, 445–447. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.V.; Keum, Y.S.; Park, S.W. Sketching the historical development of pyrimidones as the inhibitors of the HIV integrase. Eur. J. Med. Chem. 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Debnath, B.; Yu, S.; Sanchez, T.W.; Christ, F.; Liu, Y.; Debyser, Z.; Neamati, N.; Zhao, G. Design and discovery of 5-hydroxy-6-oxo-1, 6-dihydropyrimidine-4-carboxamide inhibitors of HIV-1 integrase. Bioorg. Med. Chem. 2014, 22, 5446–5453. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, J.; Salomon, C.E.; Dreis, C.D.; Vince, R. Pharmacophore and structure–activity relationships of integrase inhibition within a dual inhibitor scaffold of HIV reverse transcriptase and integrase. Bioorg. Med. Chem. 2010, 18, 4202–4211. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Maddali, K.; Dreis, C.D.; Sham, Y.Y.; Vince, R.; Pommier, Y.; Wang, Z. N-3 hydroxylation of pyrimidine-2,4-diones yields dual inhibitors of HIV reverse transcriptase and integrase. ACS Med. Chem. Lett. 2011, 2, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Maddali, K.; Dreis, C.D.; Sham, Y.Y.; Vince, R.; Pommier, Y.; Wang, Z. 6-Benzoyl-3-hydroxypyrimidine-2,4-diones as dual inhibitors of HIV reverse transcriptase and integrase. Bioorg. Med. Chem. Lett. 2011, 21, 2400–2402. [Google Scholar] [CrossRef] [PubMed]
- De Béthune, M.P. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: A review of the last 20 years (1989–2009). Antivir. Res. 2009, 85, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Arnold, E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 1. Curr. Opin. Virol. 2013, 3, 111–118. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int. J. Antimicrob. Agents 2009, 33, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Mehellou, Y.; de Clercq, E. Twenty-six years of anti-HIV drug discovery: Where do we stand and where do we go? J. Med. Chem. 2010, 53, 521–538. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. A 40-year journey in search of selective antiviral chemotherapy. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, P.; Hamasaki, T.; Isono, Y.; Sakakibara, N.; Ikejiri, M.; Maruyama, T.; Baba, M. Anti-human immunodeficiency virus type 1 activity of novel 6-substituted 1-benzyl-3-(3,5-dimethylbenzyl)uracil derivatives. Antimicrob. Agents Chemother. 2012, 56, 2581–2589. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.Y.; Isaacs, R.; Teppler, H.; Leavitt, R.Y.; Sklar, P.; Iwamoto, M.; Wenning, L.A.; Miller, M.D.; Chen, J.; Kemp, R.; et al. Raltegravir: The first HIV-1 integrasestrand transfer inhibitor in the HIV armamentarium. Ann. N. Y. Acad. Sci., 2011, 1222, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Arnold, E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 2. Curr. Opin. Virol. 2013, 3, 119–128. [Google Scholar] [CrossRef]
- Das, K.; Arnold, E.; Hughes, S. HIV-1 Reverse Transcriptase Structures. In Encyclopedia of Biological Chemistry, 2nd Ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: London, UK, 2013; Volume 2, pp. 548–553. [Google Scholar]
- Kohlstaedt, L.A.; Wang, J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 1992, 256, 1783–1790. [Google Scholar] [CrossRef] [PubMed]
- Kohlstaedt, L.A.; Wang, J.; Rice, P.A.; Friedman, J.M.; Steitz, T.A. 12 the structure of HIV-1 reverse transcriptase. Cold Spring Harb. Monogr. Arch. 1993, 23, 223–249. [Google Scholar]
- Das, K.; Lewi, P.J.; Hughes, S.H.; Arnold, E. Crystallography and the design of anti-AIDS drugs: Conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors. Prog. Biophys. Mol. Biol. 2005, 88, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, S.; Ravichandran, V.; Raman, S.; Krishnan, P.N.; Agrawal, R.K. An overview on HIV-1 reverse transcriptase inhibitors. Dig. J. Nanomater. Biostruct. 2008, 3, 171–187. [Google Scholar]
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and function of HIV-1 reverse transcriptase: Molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, X.; Meng, Q.; Wang, D.; Liu, H.; de Clercq, E.; Pannecouque, C.; Balzarini, J.; Liu, X. Novel piperidinylamino-diarylpyrimidine derivatives with dual structural conformations as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 6593–6597. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Radzio, J.; Anderson, K.S.; Sluis-Cremer, N. Probing nonnucleoside inhibitor-induced active-site distortion in HIV-1 reverse transcriptase by transient kinetic analyses. Protein Sci. 2007, 16, 1728–1737. [Google Scholar] [CrossRef] [PubMed]
- Frey, K.M.; Bollini, M.; Mislak, A.C.; Cisneros, J.A.; Gallardo-Macias, R.; Jorgensen, W.L.; Anderson, K.S. Crystal structures of HIV-1 reverse transcriptase with picomolar inhibitors reveal key interactions for drug design. J. Am. Chem. Soc. 2012, 134, 19501–19503. [Google Scholar] [CrossRef] [PubMed]
- Ekkati, A.R.; Bollini, M.; Domaoal, R.A.; Spasov, K.A.; Anderson, K.S.; Jorgensen, W.L. Discovery of dimeric inhibitors by extension into the entrance channel of HIV-1 reverse transcriptase. Bioorg. Med. Chem. Lett. 2012, 22, 1565–1568. [Google Scholar] [CrossRef] [PubMed]
- Paris, K.A.; Haq, O.; Felts, A.K.; Das, K.; Arnold, E.; Levy, R.M. Conformational landscape of the human immunodeficiency virus type 1 reverse transcriptase non-nucleoside inhibitor binding pocket: Lessons for inhibitor design from a cluster analysis of many crystal structures. J. Med. Chem. 2009, 52, 6413–6420. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Esnouf, R.; Garman, E.; Somers, D.; Ross, C.; Kirby, I.; Stammers, D. High resolution structures of HIV-1 RT from four RT–inhibitor complexes. Nat. Struct. Mol. Biol. 1995, 2, 293–302. [Google Scholar] [CrossRef]
- Esnouf, R.; Ren, J.; Ross, C.; Jones, Y.; Stammers, D.; Stuart, D. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat. Struct. Mol. Biol. 1995, 2, 303–308. [Google Scholar] [CrossRef]
- Liu, S.; Abbondanzieri, E.A.; Rausch, J.W.; Le Grice, S.F.; Zhuang, X. Slide into action: Dynamic shuttling of HIV reverse transcriptase on nucleic acid substrates. Science 2008, 322, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Radzio, J.; Sluis-Cremer, N. Efavirenz accelerates HIV-1 reverse transcriptase ribonuclease H cleavage, leading to diminished zidovudine excision. Mol. Pharmacol. 2008, 73, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Sluis-Cremer, N.; Tachedjian, G. Mechanisms of inhibition of HIV replication by non-nucleoside reverse transcriptase inhibitors. Virus Res. 2008, 134, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Bird, L.E.; Chamberlain, P.P.; Stewart-Jones, G.B.; Stuart, D.I.; Stammers, D.K. Structure of HIV-2 reverse transcriptase at 2.35-Å resolution and the mechanism of resistance to non-nucleoside inhibitors. Proc. Natl. Acad. Sci. USA 2002, 99, 14410–14415. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Kozai, S.; Demizu, Y.; Witvrouw, M.; Pannecouque, C.; Balzarini, J.; Snoeck, R.; Andrei, G.; de Clercq, E. Synthesis and anti-HIV-1 and anti-HCMV activity of 1-substituted 3-(3, 5-dimethylbenzyl) uracil derivatives. Chem. Pharm. Bull. 2006, 54, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Archer, R.H.; Dykes, C.; Gerondelis, P.; Lloyd, A.; Fay, P.; Reichman, R.C.; Bambara, R.A.; Demeter, L.M. Mutants of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase resistant to nonnucleoside reverse transcriptase inhibitors demonstrate altered rates of RNase H cleavage that correlate with HIV-1 replication fitness in cell culture. J. Virol. 2000, 74, 8390–8401. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Liu, Y.; Zheng, A.; Chen, F.; Chen, W.; de Clercq, E.; Pannacouque, C.; Balzarini, J. Design and synthesis of a new series of modified CH-diarylpyrimidines as drug-resistant HIV non-nucleoside reverse transcriptase inhibitors. Eur. J. Med. Chem. 2014, 82, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Hachiya, A.; Kodama, E.N.; Sarafianos, S.G.; Schuckmann, M.M.; Sakagami, Y.; Matsuoka, M.; Takiguchi, M.; Gatanaga1, H.; Oka, S. Amino acid mutation N348I in the connection subdomain of human immunodeficiency virus type 1 reverse transcriptase confers multiclass resistance to nucleoside and nonnucleoside reverse transcriptase inhibitors. J. Virol. 2008, 82, 3261–3270. [Google Scholar] [CrossRef] [PubMed]
- Nikolenko, G.N.; Delviks-Frankenberry, K.A.; Pathak, V.K. A novel molecular mechanism of dual resistance to nucleoside and nonnucleoside reverse transcriptase inhibitors. J. Virol. 2010, 84, 5238–5249. [Google Scholar] [CrossRef] [PubMed]
- Martins, S.; Ramos, M.J.; Fernandes, P.A. The current status of the NNRTI family of antiretrovirals used in the HAART regime against HIV infection. Curr. Med. Chem. 2008, 15, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhan, P.; Li, D.; de Clercq, E.; Liu, X. Recent advances in DAPYs and related analogues as HIV-1 NNRTIs. Curr. Med. Chem. 2011, 18, 359–376. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, W.; Zhan, P.; de Clercq, E.; Pannecouque, C.; Liu, X. Design, synthesis and anti-HIV evaluation of novel diarylnicotinamide derivatives (DANAs) targeting the entrance channel of the NNRTI binding pocket through structure-guided molecular hybridization. Eur. J. Med. Chem. 2014, 87, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Tanaka, H.; de Clercq, E.; Pauwels, R.; Balzarini, J.; Schols, D.; Nakashima, H.; Perno, C.-F.; Walker, R.T.; Miyasaka, T. Highly specific inhibition of human immunodeficiency virus type 1 by a novel 6-substituted acyclouridine derivative. Biochem. Biophys. Res. Commun. 1989, 165, 1375–1381. [Google Scholar] [CrossRef]
- Miyasaka, T.; Tanaka, H.; Baba, M.; Hayakawa, H.; Walker, R.T.; Balzarini, J.; de Clercq, E. A novel lead for specific anti-HIV-1 agents: 1-[(2-Hydroxyethoxy) methyl]-6-(phenylthio) thymine. J. Med. Chem. 1989, 32, 2507–2509. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, R.; Andries, K.; Desmyter, J.; Schols, D.; Kukla, M.J.; Breslin, H.J.; Raeymaeckers, A.; van Gelder, J.; Woestenborghs, R.; Heykants, J.; et al. Potent and selective inhibition of HIV-1 replication in vitro by a novel series of TIBO derivatives. Nature 1990, 343, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, X.; Yu, X.; Yuan, L.; Guo, Y.; Xu, W.; Liu, T.; Liu, J.; Shao, Y.; Ma, L. Inhibitory activity of 9-phenylcyclohepta[d]pyrimidinedione derivatives against different strains of HIV-1 as non-nucleoside reverse transcriptase inhibitors. J. Virol. 2011, 8, 230–246. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, J.; Huang, Y.; Wang, R.; Zhang, L.; Qiao, K.; Li, L.; Liu, C.; Ouyang, Y.; Xu, W.; et al. Design, synthesis, and biological evaluation of 1-[(2-benzyloxyl/alkoxyl)methyl]-5-halo-6-aryluracils as potent HIV-1 non-nucleoside reverse transcriptase inhibitors with an improved drug resistance profile. J. Med. Chem. 2012, 55, 2242–2250. [Google Scholar] [CrossRef] [PubMed]
- Buckheit, R.W., Jr.; Hartman, T.L.; Watson, K.M.; Chung, S.G.; Cho, E.H. Comparative evaluation of the inhibitory activities of a series of pyrimidinedione congeners that inhibit human immunodeficiency virus types 1 and 2. Antimicrob. Agents Chemother. 2008, 52, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Buckheit, K.W.; Yang, L.; Buckheit, R.W., Jr. Development of dual-acting pyrimidinediones as novel and highly potent topical anti-HIV microbicides. Antimicrob. Agents Chemother. 2011, 55, 5243–5254. [Google Scholar] [CrossRef] [PubMed]
- Hartman, T.L.; Yang, L.; Buckheit, R.W., Jr. Antiviral interactions of combinations of highly potent 2,4 (1H, 3H)-pyrimidinedione congeners and other anti-HIV agents. Antivir. Res. 2011, 92, 505–508. [Google Scholar]
- Ham, A.S.; Rohan, L.C.; Boczar, A.; Yang, L.; Buckheit, K.W.; Buckheit, R.W., Jr. Vaginal film drug delivery of the pyrimidinedione IQP-0528 for the prevention of HIV infection. Pharm. Res. 2012, 29, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Ham, A.S.; Lustig, W.; Yang, L.; Boczar, A.; Buckheit, K.W.; Buckheit, R.W., Jr. In vitro and ex vivo evaluations on transdermal delivery of the HIV inhibitor IQP-0410. PLoS ONE 2013, 8, e75306. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, A.; Simmons, A.P.; Ugaonkar, S.R.; Watson, K.M.; Dezzutti, C.S.; Rohan, L.C.; Buckheit, R.W., Jr.; Kiser, P.F. Vaginal microbicide gel for delivery of IQP-0528, a pyrimidinedione analog with a dual mechanism of action against HIV-1. Antimicrob. Agents Chemother. 2011, 55, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; He, Q.Q.; Yang, S.Q.; Ma, X.D.; Chen, F.E.; de Clercq, E.; Balzarini, J.; Pannecouque, C. Synthesis and structure–activity relationship of novel diarylpyrimidines with hydromethyl linker (CH(OH)-DAPYs) as HIV-1 NNRTIs. Bioorg. Med. Chem. 2011, 19, 5117–5124. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.Q.; Zeng, Z.S.; Liang, Y.H.; Chen, F.E.; Pannecouque, C.; Balzarini, J.; de Clercq, E. Synthesis and biological evaluation of 4-(hydroxyimino) arylmethyl diarylpyrimidine analogues as potential non-nucleoside reverse transcriptase inhibitors against HIV. Bioorg. Med. Chem. 2010, 18, 2370–2374. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; Li, Z.M.; Ma, X.D.; Yang, S.Q.; He, Q.Q.; Chen, F.E.; de Clercq, E.; Balzarini, J.; Pannecouque, C. Chiral resolution, absolute configuration assignment and biological activity of racemic diarylpyrimidine CH(OH)-DAPY as potent non-nucleoside HIV-1 reverse transcriptase inhibitors. Eur. J. Med. Chem. 2012, 53, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.H.; Huang, X.Y.; Wu, H.Q.; Chen, W.X.; He, Q.Q.; Chen, F.E.; de Clercq, E.; Pannecouque, C. Structural modifications of CH(OH)-DAPYs as new HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. 2014, 22, 2535–2541. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.Q.; Liang, Y.H.; Zeng, Z.S.; Chen, F.E.; Balzarini, J.; Pannecouque, C.; de Clercq, E. Structural modifications of DAPY analogues with potent anti-HIV-1 activity. ChemMedChem 2009, 4, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Pannecouque, C.; Daelemans, D.; Ma, X.D.; Liu, Y.; Chen, F.E.; de Clercq, E. Molecular design, synthesis and biological evaluation of BP-O-DAPY and O-DAPY derivatives as non-nucleoside HIV-1 reverse transcriptase inhibitors. Eur. J. Med. Chem. 2013, 65, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.S.; Liang, Y.H.; Feng, X.Q.; Chen, F.E.; Pannecouque, C.; Balzarini, J.; de Clercq, E. Lead optimization of diarylpyrimidines as non-nucleoside inhibitors of HIV-1 reverse transcriptase. ChemMedChem 2010, 5, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; Yang, S.Q.; He, Q.Q.; Ma, X.D.; Chen, F.E.; Dai, H.F.; de Clercq, E.; Balzarini, J.; Pannecouque, C. Design, synthesis and biological evaluation of cycloalkyl arylpyrimidines (CAPYs) as HIV-1 NNRTIs. Bioorg. Med. Chem. 2011, 19, 7093–7099. [Google Scholar] [CrossRef] [PubMed]
- D’Cruz, O.J.; Uckun, F.M. Dawn of non-nucleoside inhibitor-based anti-HIV microbicides. J. Antimicrob. Chemother. 2006, 57, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Radi, M.; Pagano, M.; Franchi, L.; Castagnolo, D.; Schenone, S.; Casaluce, G.; Zamperini, C.; Dreassi, E.; Maga, G.; Samuele, A.; et al. Synthesis, biological activity, and ADME properties of novel S-DABOs/N-DABOs as HIV reverse transcriptase inhibitors. ChemMedChem 2012, 7, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Rotili, D.; Tarantino, D.; Artico, M.; Nawrozkij, M.B.; Gonzalez-Ortega, E.; Clotet, B.; Samuele, A.; Esté, J.A.; Maga, G.; Mai, A. Diarylpyrimidine-dihydrobenzyloxopyrimidine hybrids: New, wide-spectrum anti-HIV-1 agents active at (sub)-nanomolar level. J. Med. Chem. 2011, 54, 3091–3096. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Chen, F.E.; de Clercq, E. Dihydro-alkoxyl-benzyl-oxopyrimidine derivatives (DABOs) as non-nucleoside reverse transcriptase inhibitors: An update review (2001–2011). Curr. Med. Chem. 2012, 19, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Q.; Yan, Z.H.; Chen, W.X.; He, Q.Q.; Chen, F.E.; de Clercq, E.; Balzarini, J.; Daelemans, D.; Pannecouque, C. Towards new C6-rigid S-DABO HIV-1 reverse transcriptase inhibitors: Synthesis, biological investigation and molecular modeling studies. Bioorg. Med. Chem. 2013, 21, 6477–6483. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Chen, F.E.; de Clercq, E.; Balzarini, J.; Pannecouque, C. Synthesis and in vitro anti-HIV evaluation of a new series of 6-arylmethyl-substituted S-DABOs as potential non-nucleoside HIV-1 reverse transcriptase inhibitors. Eur. J. Med. Chem. 2009, 44, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Liu, C.; Guo, Y.; Wang, R.; Zhang, J.; Ma, L.; Zhang, Z.; Wang, X.; Cui, Y.; Liu, J. Synthesis and biological evaluation of novel C5 halogen-functionalized S-DABO as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. 2010, 18, 3231–3237. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Liu, C.; Zhang, J.; Guo, Y.; Zhang, S.; Zhang, Z.; Wang, X.; Zhang, L.; Liu, J. Synthesis and biological evaluation of novel 2-arylalkylthio-4-amino-6-benzyl pyrimidines as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3003–3005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tang, X.; Cao, Y.; Wu, S.; Zhang, Y.; Zhao, J.; Guo, Y.; Tian, C.; Zhang, Z.; Liu, J.; et al. Synthesis and biological evaluation of novel 2-arylalkylthio-5-iodine-6-substituted-benzyl-pyrimidine-4 (3H)-ones as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Molecules 2014, 19, 7104–7121. [Google Scholar] [CrossRef] [PubMed]
- Bollini, M.; Domaoal, R.A.; Thakur, V.V.; Gallardo-Macias, R.; Spasov, K.A.; Anderson, K.S.; Jorgensen, W.L. Computationally-guided optimization of a docking hit to yield catechol diethers as potent anti-HIV agents. J. Med. Chem. 2011, 54, 8582–8591. [Google Scholar] [CrossRef] [PubMed]
- Frey, K.M.; Puleo, D.E.; Spasov, K.A.; Bollini, M.; Jorgensen, W.L.; Anderson, K.S. Structure-based evaluation of non-nucleoside inhibitors with improved potency and solubility that target HIV reverse transcriptase variants. J. Med. Chem. 2015, 58, 2737–2745. [Google Scholar] [CrossRef] [PubMed]
- Rai, D.; Chen, W.; Tian, Y.; Chen, X.; Zhan, P.; de Clercq, E.; Pannecouque, C.; Balzarini, J.; Liu, X. Design, synthesis and biological evaluation of 3-benzyloxy-linked pyrimidinylphenylamine derivatives as potent HIV-1 NNRTIs. Bioorg. Med. Chem. 2013, 21, 7398–7405. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Kozai, S.; Yamasaki, T.; Witvrouw, M.; Pannecouque, C.; Balzarini, J.; Snoeck, R.; Andrei, G.; de Clercq, E. Synthesis and antiviral activity of 1,3-disubstituted uracils against HIV-1 and HCMV. Antivir. Chem. Chemother. 2003, 14, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Kozai, S.; Demizu, Y.; Witvrouw, M.; Pannecouque, C.; Balzarini, J.; Snoeck, R.; Andrei, G.; de Clercq, E. Synthesis and Antiviral Activity of 1-Substituted 3-(3, 5-dimethylbenzyl) Uracil against HIV-1. In Nucleic Acids Symposium Series; Oxford University Press: Oxford, UK, 2004; Volume 48, pp. 3–4. [Google Scholar]
- Isono, Y.; Sakakibara, N.; Ordonez, P.; Hamasaki, T.; Baba, M.; Ikejiri, M.; Maruyama, T. Synthesis of 1-benzyl-3-(3, 5-dimethylbenzyl) uracil derivatives with potential anti-HIV activity. Antivir. Chem. Chemother. 2011, 22, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Hamasaki, T.; Baba, M.; Demizu, Y.; Kurihara, M.; Irie, K.; Asada, E.; Kato, Y.; Maruyama, T. Synthesis and evaluation of novel 3-(3, 5-dimethylbenzyl) uracil analogs as potential anti-HIV-1 agents. Bioorg. Med. Chem. 2013, 21, 5900–5906. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Baba, M.; Okamoto, M.; Toyama, M.; Demizu, Y.; Misawa, T.; Kurihara, M.; Irie, K.; Kato, Y.; Maruyama, T. Design, synthesis, and anti-HIV-1 activity of 1-aromatic methyl-substituted 3-(3, 5-dimethylbenzyl) uracil and N-3, 5-dimethylbenzyl-substituted urea derivatives. Antivir. Chem. Chemother. 2015, 24, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Novikov, M.S.; Buckheit, R.W.; Temburnikar, K.; Khandazhinskaya, A.L.; Ivanov, A.V.; Seley-Radtke, K.L. 1-Benzyl derivatives of 5-(arylamino)uracils as anti-HIV-1 and anti-EBV agents. Bioorg. Med. Chem. 2010, 18, 8310–8314. [Google Scholar] [CrossRef] [PubMed]
- Novikov, M.S.; Ivanova, O.N.; Ivanov, A.V.; Ozerov, A.A.; Valuev-Elliston, V.T.; Temburnikar, K.; Pannecouque, C.; Balzarini, J.; Seley-Radtke, K.L. 1-[2-(2-Benzoyl-and 2-benzylphenoxy) ethyl] uracils as potent anti-HIV-1 agents. Bioorg. Med. Chem. 2011, 19, 5794–5802. [Google Scholar] [CrossRef] [PubMed]
- Novikov, M.S.; Valuev-Elliston, V.T.; Babkov, D.A.; Paramonova, M.P.; Ivanov, A.V.; Gavryushov, S.A.; Pannecouque, C.; Balzarini, J.; Snoeck, R.; Andrei, G.; et al. N1,N3-disubstituted uracils as nonnucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg. Med. Chem. 2013, 21, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Matyugina, E.S.; Valuev-Elliston, V.T.; Geisman, A.N.; Novikov, M.S.; Chizhov, A.O.; Kochetkov, S.N.; Seley-Radtke, K.L.; Khandazhinskaya, A.L. Structure-activity evaluation of new uracil-based non-nucleoside inhibitors of HIV reverse transcriptase. MedChemComm 2013, 4, 1443–1451. [Google Scholar] [CrossRef]
- Chemical Identifier Resolver beta 4. Available online: http://cactus.nci.nih.gov/chemical/structure (accessed on 23 February 2013).
- Putz, M.V.; Dudaş, N.A. Variational principles for mechanistic quantitative structure–activity relationship (QSAR) studies: Application on uracil derivatives’ anti-HIV action. Struct. Chem. 2013, 24, 1873–1893. [Google Scholar] [CrossRef]
- Putz, M.V.; Dudaş, N.A. Determining chemical reactivity driving biological activity from SMILES transformations: The bonding mechanism of anti-HIV pyrimidines. Molecules 2013, 18, 9061–9116. [Google Scholar] [CrossRef] [PubMed]
- Putz, M.V. Quantum Nanochemistry. A Fully Integrated Approach: Vol V. Quantum Structure-Activity Relationship (Qu-SAR); Apple Academic Press & CRC Press: Oakville, ON, Canada; Toronto, ON, Canada; Waretown, NJ, USA, 2015. [Google Scholar]
- Moldoveanu, C.C.; Jones, P.G.; Mangalagiu, I.I. Spiroheterocyclic compounds: Old stories with new outcomes. Tetrahedron Lett. 2009, 50, 7205–7208. [Google Scholar] [CrossRef]
- Gammon, D.B.; Snoeck, R.; Fiten, P.; Krecmerova, M.; Holy, A.; de Clercq, E.; Opdenakker, G.; Evans, D.H.; Andrei, G. Mechanism of antiviral drug resistance of Vaccinia virus: Identification of residues in the viral DNA polymerase conferring differential resistance to antipoxvirus drugs. J. Virol. 2008, 82, 12520–12534. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.Y.; Zheng, Z.B.; Mi, C.L.; Zhou, X.B.; Yan, H.; Gong, Z.H.; Li, S. Synthesis and evaluation of novel chloropyridazine derivatives as potent human rhinovirus (HRV) capsid-binding inhibitors. Bioorg. Med. Chem. 2009, 17, 621–624. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. New approaches toward anti-HIV chemotherapy. J. Med. Chem. 2005, 48, 1297–1313. [Google Scholar] [CrossRef] [PubMed]
- Muhanji, C.I.; Hunter, R. Current developments in the synthesis and biological activity of HIV-1 double-drug inhibitors. Curr. Med. Chem. 2007, 14, 1207–1222. [Google Scholar] [CrossRef] [PubMed]
- Butnariu, R.; Caprosu, M.; Bejan, V.; Ungureanu, M.; Poiata, A.; Tuchilus, C.; Florescu, M.; Mangalagiu, I.I. Pyridazine and phthalazine derivatives with potential antimicrobial activity. J. Heterocycl. Chem. 2007, 44, 1149–1152. [Google Scholar] [CrossRef]
- Butnariu, R.; Mangalagiu, I.I. New pyridazine derivatives: Synthesis, chemistry and biological activity. Bioorg. Med. Chem. 2009, 17, 2823–2829. [Google Scholar] [CrossRef] [PubMed]
- Balan, A.M.; Florea, O.; Moldoveanu, C.; Zbancioc, G.; Iurea, D.; Mangalagiu, I.I. Diazinium salts with dihydroxyacetophenone skeleton: Syntheses and antimicrobial activity. Eur. J. Med. Chem. 2009, 44, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- Najahi, E.; Vanthuyne, N.; Nepveu, F.; Jean, M.; Alkorta, I.; Elguero, J.; Roussel, C. Atropisomerization in N-aryl-2(1H)-pyrimidin-(thi) ones: A ring-opening/rotation/ring-closure process in place of a classical rotation around the pivot bond. J. Org. Chem. 2013, 78, 12577–12584. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P.; Cassani, S.; Chirico, N. QSARINS-Chem: Insubria datasets and new QSAR/QSPR models for environmental pollutants in QSARINS. J. Comp. Chem. 2014, 35, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Bauman, J.D.; Clark, A.D.; Frenkel, Y.V.; Lewi, P.J.; Shatkin, A.J.; Hughes, S.H.; Arnold, E. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: Strategic flexibility explains potency against resistance mutations. Proc. Natl. Acad. Sci. USA 2008, 105, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, J.; Sigurðsson, S.; Löwgren, S.; O Andersson, H.; Sahlberg, C.; Noréen, R.; Fridborg, K.; Zhang, H.; Unge, T. Structural basis for the inhibitory efficacy of efavirenz (DMP-266), MSC194 and PNU142721 towards the HIV-1 RT K103N mutant. Eur. J. Biochem. 2002, 269, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comp. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H. J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comp. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins Struct. Funct. Bioinform. 2007, 69, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Firoz, A.; Malik, A.; Afzal, O.; Jha, V. ContPro: A web tool for calculating amino acid contact distances in protein from 3D–structure at different distance threshold. Bioinformation 2010, 5, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Rong, J.; Zhang, B.; Hu, L.; Wang, X.; Zeng, C. Design and synthesis of N-methylpyrimidone derivatives as HIV-1 integrase inhibitors. Bioorg. Med. Chem. 2015, 23, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Li, Y.; Hao, M.; Zhang, S.; Ai, C. Docking, molecular dynamics and quantitative structure-activity relationship studies for HEPTs and DABOs as HIV-1 reverse transcriptase inhibitors. J. Mol. Model. 2012, 18, 2185–2198. [Google Scholar] [CrossRef] [PubMed]
- Lansdon, E.B.; Brendza, K.M.; Hung, M.; Wang, R.; Mukund, S.; Jin, D.; Birkus, G.; Kutty, N.; Liu, X. Crystal structures of HIV-1 reverse transcriptase with etravirine (TMC125) and rilpivirine (TMC278): Implications for drug design. J. Med. Chem. 2010, 53, 4295–4299. [Google Scholar] [CrossRef] [PubMed]
- Lewin, S.R.; Evans, V.A.; Elliott, J.H.; Spire, B.; Chomont, N. Finding a cure for HIV: Will it ever be achievable? J. Int. AIDS Soc. 2011, 24, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Fauci, A.S. HIV reservoirs: Pathogenesis and obstacles to viral eradication and cure. AIDS 2012, 26, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.; Barré-Sinoussi, F. Controversies in HIV cure research. J. Int. AIDS Soc. 2012, 15, 16–28. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Putz, M.V.; Dudaș, N.A.; Isvoran, A. Double Variational Binding—(SMILES) Conformational Analysis by Docking Mechanisms for Anti-HIV Pyrimidine Ligands. Int. J. Mol. Sci. 2015, 16, 19553-19601. https://doi.org/10.3390/ijms160819553
Putz MV, Dudaș NA, Isvoran A. Double Variational Binding—(SMILES) Conformational Analysis by Docking Mechanisms for Anti-HIV Pyrimidine Ligands. International Journal of Molecular Sciences. 2015; 16(8):19553-19601. https://doi.org/10.3390/ijms160819553
Chicago/Turabian StylePutz, Mihai V., Nicoleta A. Dudaș, and Adriana Isvoran. 2015. "Double Variational Binding—(SMILES) Conformational Analysis by Docking Mechanisms for Anti-HIV Pyrimidine Ligands" International Journal of Molecular Sciences 16, no. 8: 19553-19601. https://doi.org/10.3390/ijms160819553
APA StylePutz, M. V., Dudaș, N. A., & Isvoran, A. (2015). Double Variational Binding—(SMILES) Conformational Analysis by Docking Mechanisms for Anti-HIV Pyrimidine Ligands. International Journal of Molecular Sciences, 16(8), 19553-19601. https://doi.org/10.3390/ijms160819553