
Insight into the Structural Determinants of Imidazole Scaffold-Based Derivatives as TNF-α Release Inhibitors by in Silico Explorations

Abstract
:
1. Introduction
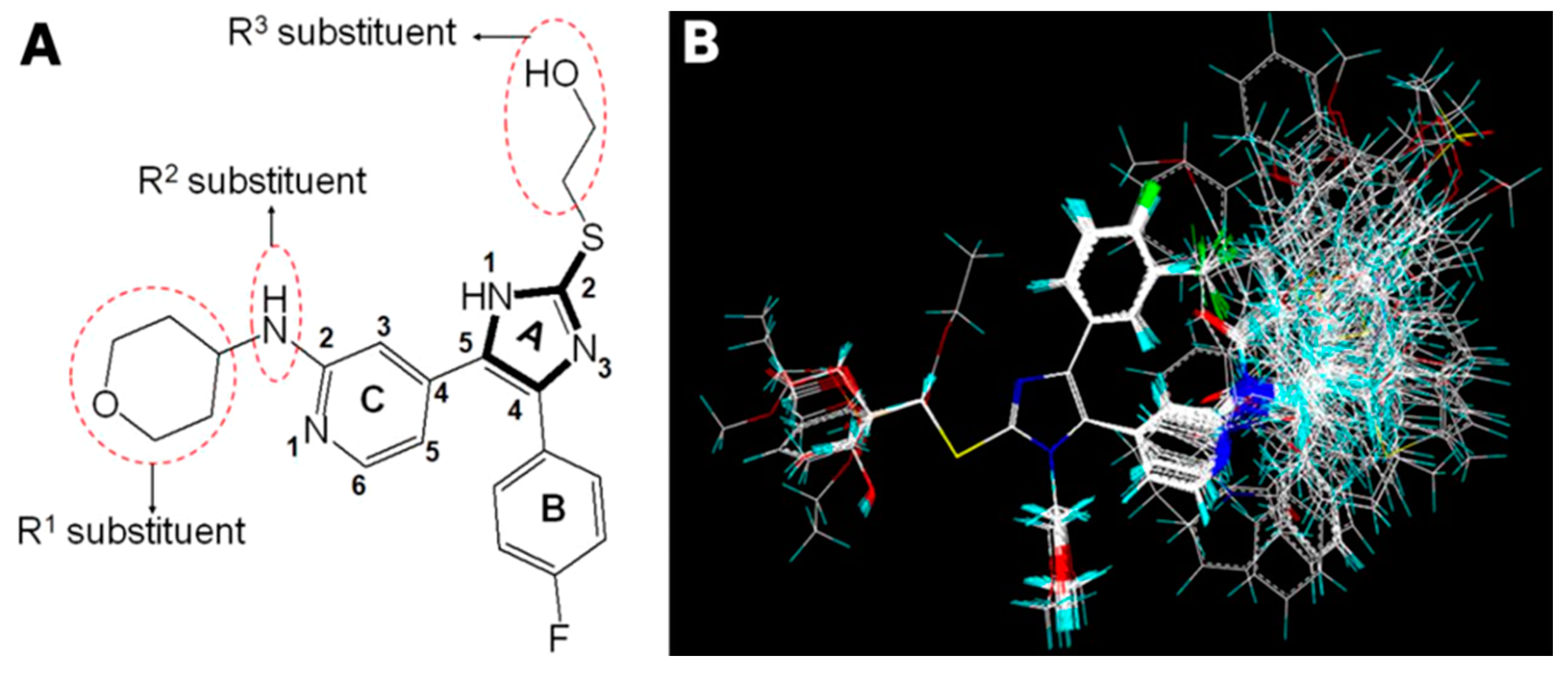
2. Results and Discussion
2.1. Split the Training and Test Sets

2.2. 3D-QSAR Statistical Results


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLS Statistics | Model A | Model B | ||
|---|---|---|---|---|
| CoMFA | CoMSIA | CoMFA | CoMSIA | |
| Q2 | 0.524 | 0.593 | 0.557 | 0.598 |
| R2ncv | 0.856 | 0.778 | 0.740 | 0.767 |
| SEE | 0.323 | 0.399 | 0.432 | 0.409 |
| F | 133.655 | 99.117 | 80.388 | 93.091 |
| R2pre | 0.730 | 0.876 | 0.748 | 0.860 |
| SEP | 0.588 | 0.541 | 0.564 | 0.538 |
| OPN | 5 | 4 | 4 | 4 |
| Contribution | ||||
| S | 0.574 | 0.121 | 0.492 | 0.135 |
| E | 0.426 | 0.294 | 0.373 | 0.279 |
| H | - | 0.195 | - | 0.158 |
| D | - | 0.208 | - | 0.195 |
| A | - | 0.181 | - | 0.146 |
| ClogP | - | - | 0.135 | 0.086 |
2.2.1. CoMFA Details
2.2.2. CoMSIA Details
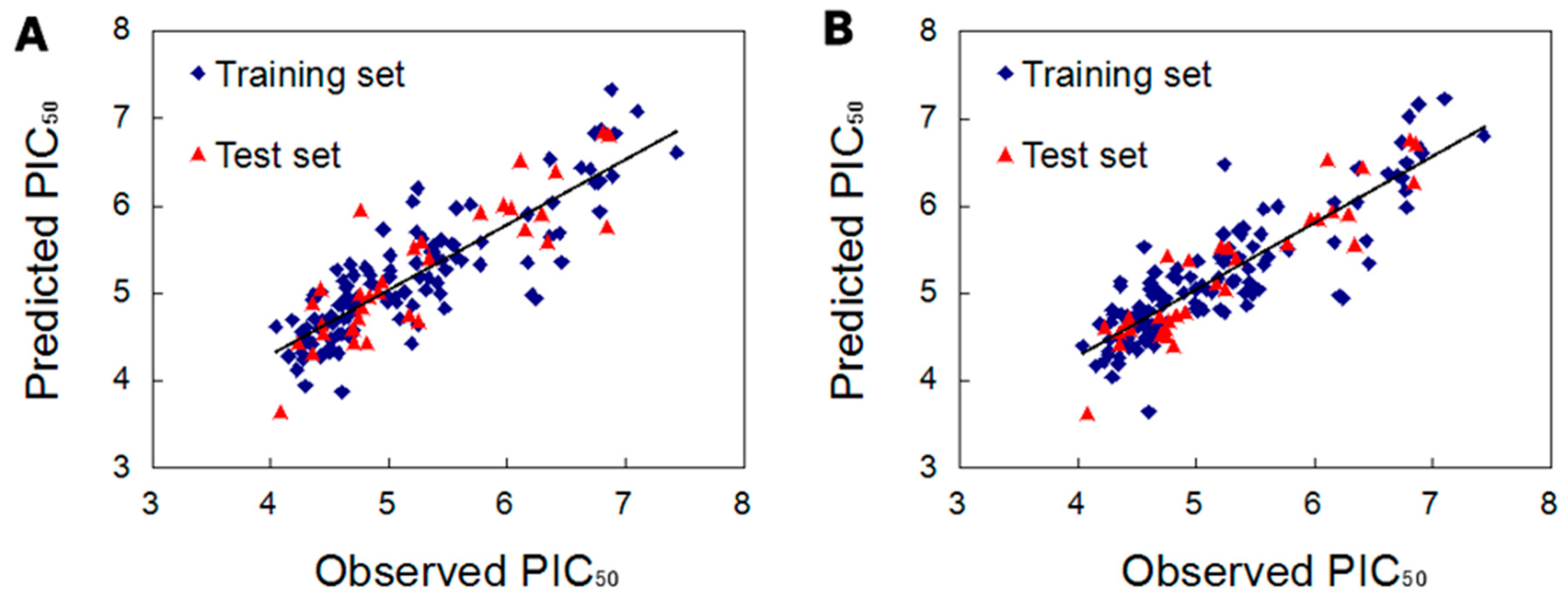
2.2.3. Validation of the 3D QSAR Models

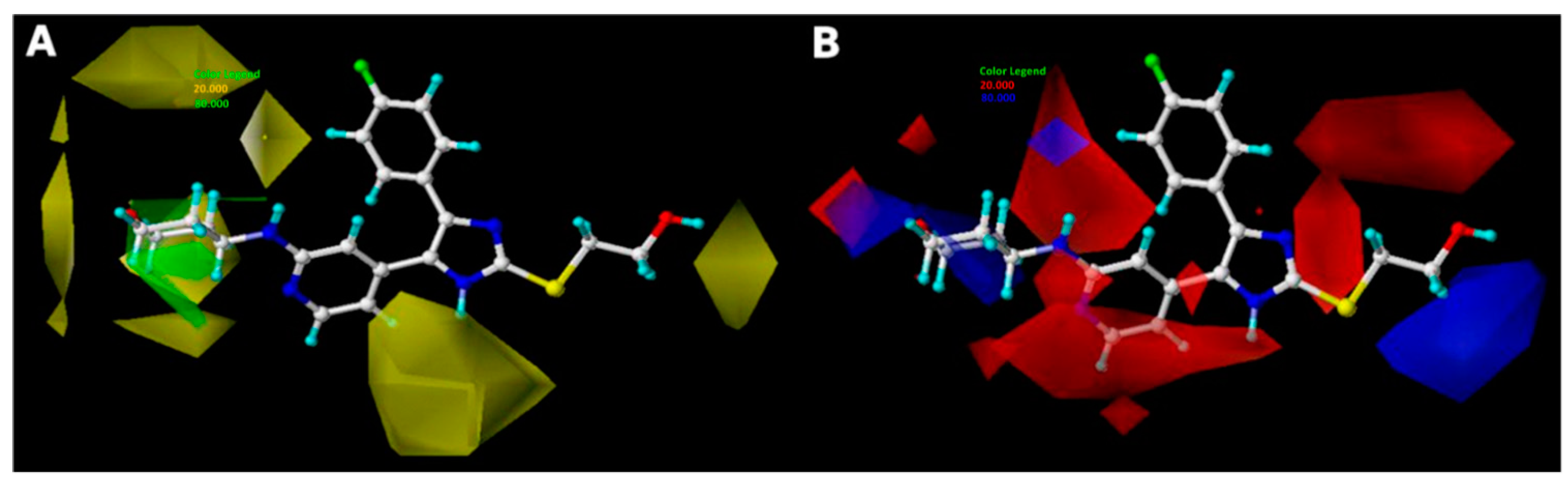
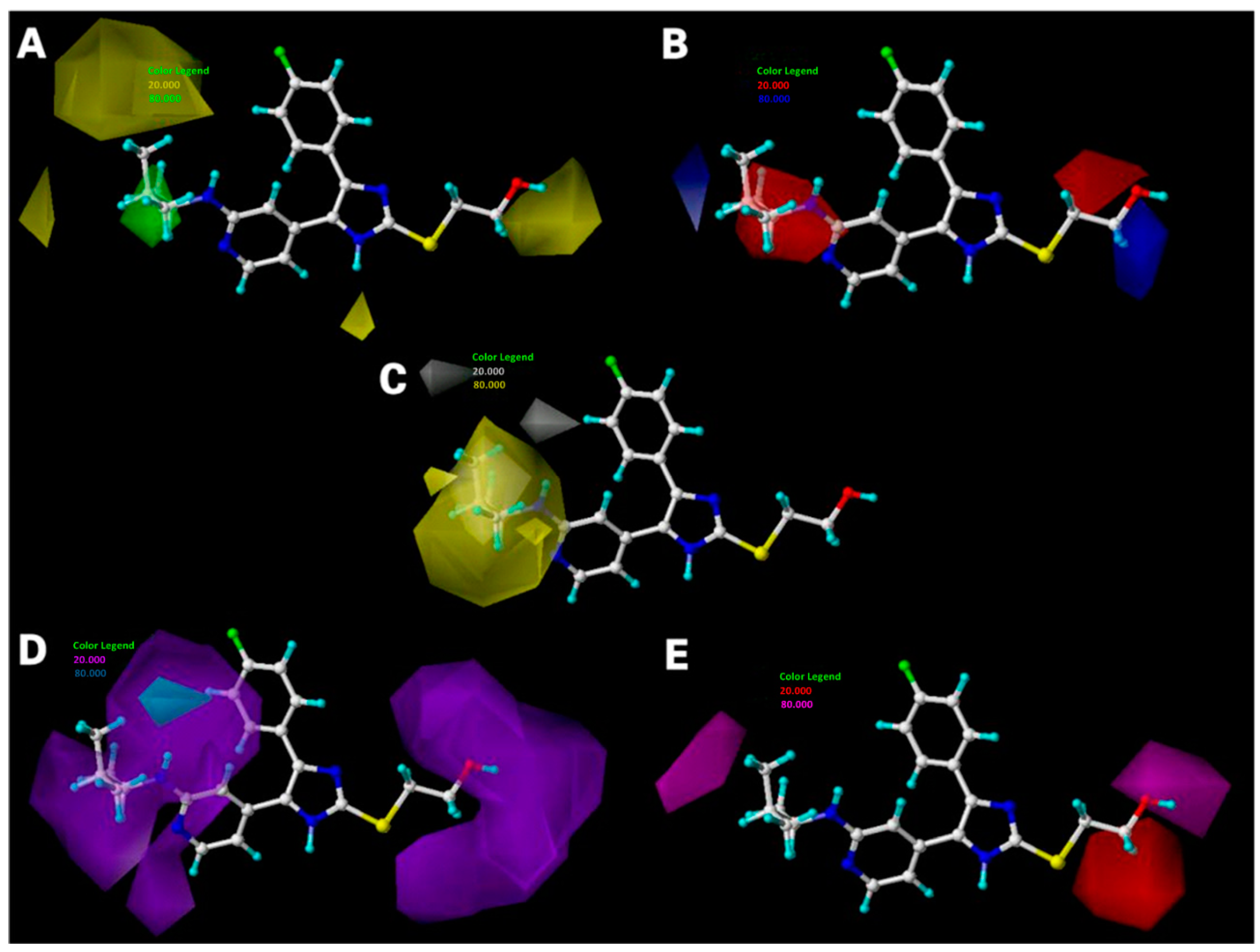
2.3. Interpretation of 3D-QSAR Contour Maps


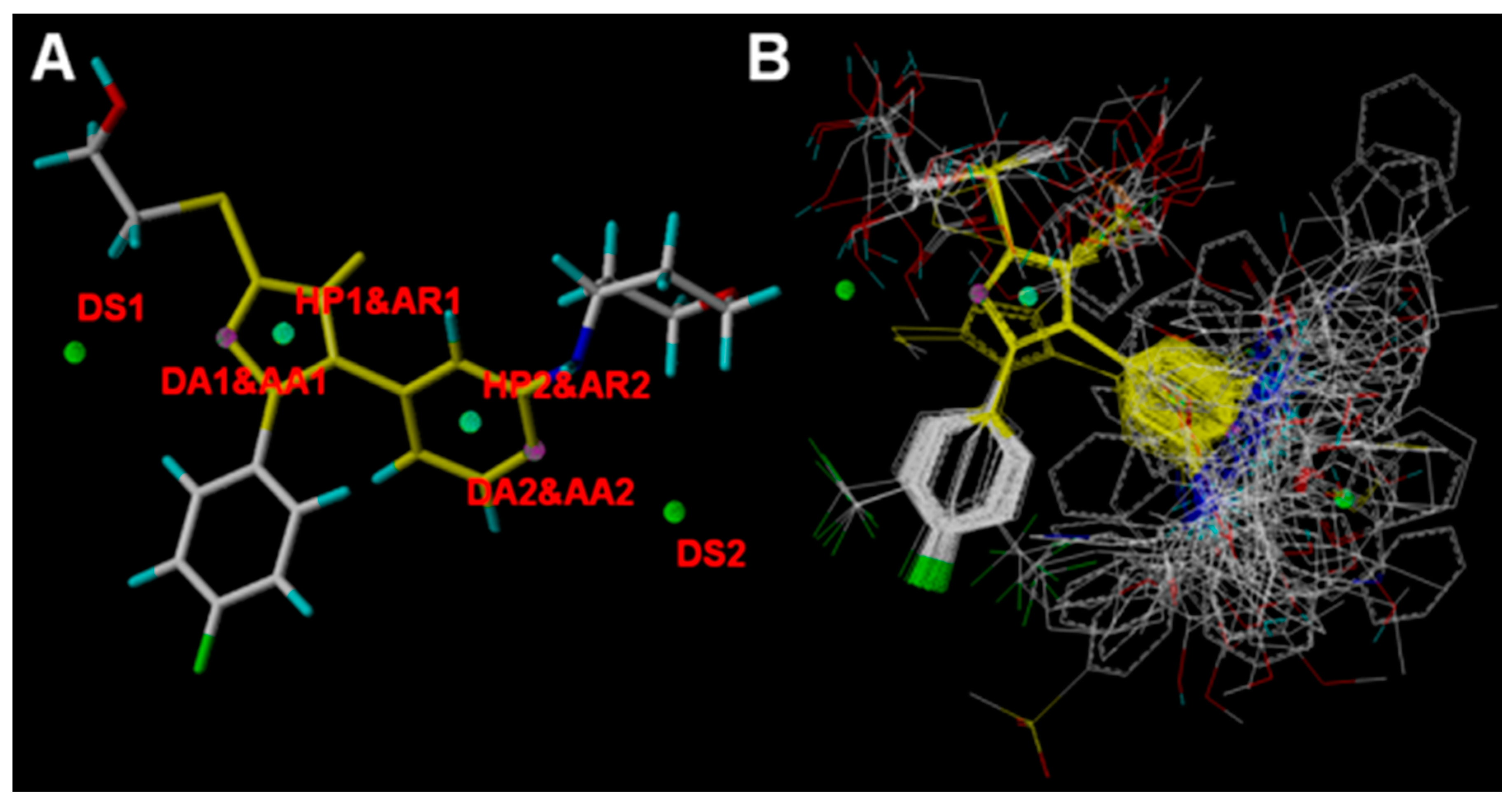
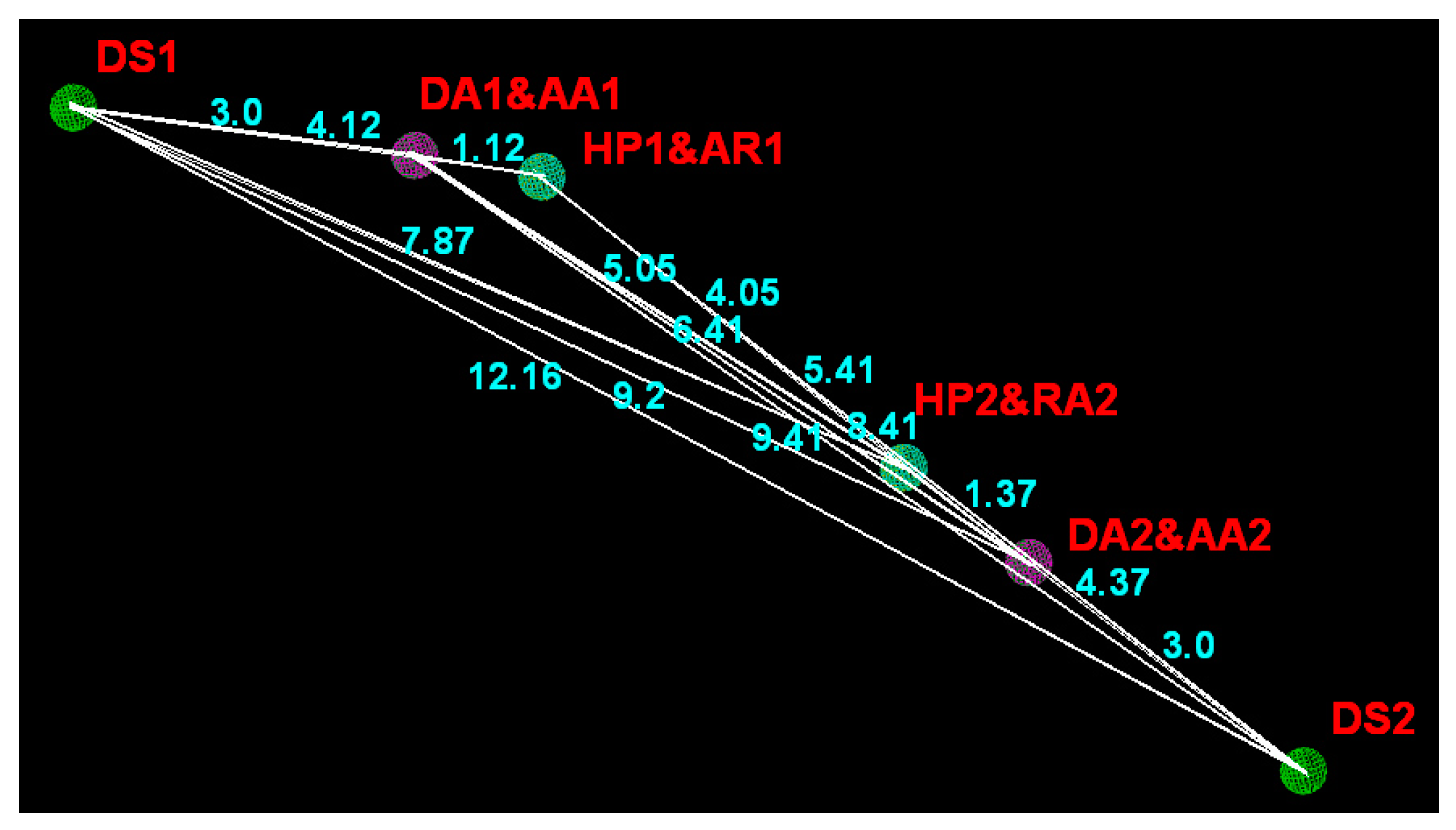
2.4. Pharmacophore Modeling
| Model | Size a | Hits b | Score c | Tolerance d | Dmean e |
|---|---|---|---|---|---|
| MODEL_006 | 10 | 100 | 4.1075 | 0.25 | 4.5615 |
| MODEL_003 | 10 | 100 | 4.1015 | 0.50 | 4.5264 |
| MODEL_008 | 10 | 100 | 4.1015 | 0.50 | 4.5263 |
| MODEL_007 | 10 | 100 | 3.4537 | 0.50 | 3.7971 |
| MODEL_005 | 10 | 100 | 3.4526 | 0.25 | 3.7925 |
| MODEL_001 | 10 | 100 | 3.4517 | 0.25 | 3.789 |
| MODEL_004 | 10 | 100 | 3.4517 | 0.25 | 3.789 |
| MODEL_002 | 10 | 100 | 3.4516 | 0.25 | 3.7885 |


| Domain | DS1 | DA1/AA1 | HP1/AR1 | HP2/AR2 | AA2/DA2 |
|---|---|---|---|---|---|
| DA1/AA1 | 3.00 ± 0.25 | – | – | – | – |
| HP1/AR1 | 4.12 ± 0.25 | 1.12 ± 0.25 | – | – | – |
| HP2/AR2 | 7.87 ± 0.25 | 5.05 ± 0.25 | 4.05 ± 0.25 | – | – |
| AA2/DA2 | 9.20 ± 0.25 | 6.41 ± 0.25 | 5.41 ± 0.25 | 1.37 ± 0.25 | – |
| DS2 | 12.16 ± 0.25 | 9.41 ± 0.25 | 8.41 ± 0.25 | 4.37 ± 0.25 | 3.00 ± 0.25 |
3. Experimental Section
3.1. Dataset and Biological Activity
3.2. Molecular Modeling and Alignment Procedure
3.3. CoMFA and CoMSIA
3.4. Partial Least Square Analysis and Statistical Validation
3.5. DISCOtech Analysis
4. Conclusions
- Bulky substituents at R1 position may improve the inhibitory activity.
- Hydrophobic groups around R1 substituent are helpful to enhance the potency of the inhibitors.
- Electropositive groups at R3 position are beneficial to improve the biological activity of inhibitors.
- H-bond donor groups around ring C and acceptor groups at R3 substituent promote the inhibitory activity, respectively.
- Hydrophobic interaction and hydrogen bonds were the crucial factors acting on the inhibitory activity of TNF-α release.

Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Karin, M. Mitogen activated protein kinases as targets for development of novel anti-inflammatory drugs. Anna. Rheum. Dis. 2004, 63, ii62–ii64. [Google Scholar] [CrossRef] [PubMed]
- Laufer, S.A.; Hauser, D.R.; Domeyer, D.M.; Kinkel, K.; Liedtke, A.J. Design, synthesis, and biological evaluation of novel Tri- and tetrasubstituted imidazoles as highly potent and specific ATP-mimetic inhibitors of p38 MAP kinase: Focus on optimized interactions with the enzyme’s surface-exposed front region. J. Med. Chem. 2008, 51, 4122–4149. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J.; Cerami, A. Tumor necrosis factor, other cytokines and disease. Ann. Rev. Cell Boil. 1993, 9, 317–343. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Inflammatory cytokines: Interleukin-1 and tumor necrosis factor as effector molecules in autoimmune diseases. Curr. Opin. Immunol. 1991, 3, 941–948. [Google Scholar] [CrossRef]
- Elliott, M.J.; Maini, R.N.; Feldmann, M.; Kalden, J.R.; Antoni, C.; Smolen, J.S.; Leeb, B.; Breedveld, F.C.; Macfarlane, J.D.; Bijl, H.; et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor α (cA2) versus placebo in rheumatoid arthritis. The Lancet 1994, 344, 1105–1110. [Google Scholar] [CrossRef]
- Elliott, M.J.; Maini, R.N.; Feldmann, M.; Long-Fox, A.; Charles, P.; Bijl, H.; Woody, J.N. Repeated therapy with monoclonal antibody to tumour necrosis factor α (cA2) in patients with rheumatoid arthritis. The Lancet 1994, 344, 1125–1127. [Google Scholar] [CrossRef]
- Rankin, E.C.; Choy, E.H.; Kassimos, D.; Kingsley, G.H.; Sopwith, A.M.; Isenberg, D.A.; Panayi, G.S. The therapeutic effects of an engineered human anti-tumour necrosis factor α antibody (CDP571) in rheumatoid arthritis. Brit. J. Rheumatol. 1995, 34, 334–342. [Google Scholar] [CrossRef]
- Van Dullemen, H.M.; van Deventer, S.J.; Hommes, D.W.; Bijl, H.A.; Jansen, J.; Tytgat, G.N.; Woody, J. Treatment of Crohn’s disease with anti-tumor necrosis factor chimeric monoclonal antibody (cA2). Gastroenterology 1995, 109, 129–135. [Google Scholar] [CrossRef]
- Moreland, L.W.; Baumgartner, S.W.; Schiff, M.H.; Tindall, E.A.; Fleischmann, R.M.; Weaver, A.L.; Ettlinger, R.E.; Cohen, S.; Koopman, W.J.; Mohler, K.; et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N. Engl. J. Med. 1997, 337, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Pierre, K.; Christiane, B.; Hartmut, J.; Stefan, L. Targeting the ribose and phosphate binding site of p38 mitogen-activated protein (MAP) kinase: Synthesis and biological testing of 2-alkylsulfanyl-, 4(5)-aryl-, 5(4)-heteroaryl-substituted imidazoles. J. Med. Chem. 2008, 51, 5630–5640. [Google Scholar]
- Waetzig, G.H.; Schreiber, S. Review article: Mitogen-activated protein kinases in chronic intestinal inflammation—Targeting ancient pathways to treat modern diseases. Aliment. Pharmacol. Ther. 2003, 18, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Fukami, Y.; Tokmakov, A.A.; Konaka, K.; Sato, K. Peptide inhibitors of the mitogen-activated protein kinase pathway: A structure-mimetic peptide corresponding to the conserved inter-DFG-APE region in the kinase domain. Pharmacol. Ther. 1999, 82, 399–407. [Google Scholar] [CrossRef]
- Engelberg, D. Stress-activated protein kinases-tumor suppressors or tumor initiators? Semin. Cancer Biol. 2004, 14, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Dalrymple, S.A. p38 mitogen activated protein kinase as a therapeutic target for Alzheimer’s disease. J. Mol. Neurosci. 2002, 19, 295–299. [Google Scholar] [CrossRef]
- Feng, Y.J.; Li, Y.Y. The role of p38 mitogen-activated protein kinase in the pathogenesis of inflammatory bowel disease. J. Dig. Dis. 2011, 12, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.; Laufer, S. Small molecular anti-cytokine agents. Med. Res. Rev. 2006, 26, 1–62. [Google Scholar] [CrossRef] [PubMed]
- Saklatvala, J. The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Curr. Opin. Pharmacol. 2004, 4, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Bracht, C.; Hauser, D.R.; Schattel, V.; Albrecht, W.; Laufer, S.A. Synthesis and biological testing of N-aminoimidazole-based p38α MAP kinase inhibitors. Chemmedchem 2010, 5, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, K.; Hauser, D.R.; Unger, A.; Albrecht, W.; Laufer, S.A. 2-Acylaminopyridin-4-ylimidazoles as p38 MAP kinase inhibitors: Design, synthesis, and biological and metabolic evaluations. Chemmedchem 2009, 4, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Ravindra, G.K.; Achaiah, G.; Sastry, G.N. Molecular modeling studies of phenoxypyrimidinyl imidazoles as p38 kinase inhibitors using QSAR and docking. Eur. J. Med. Chem. 2008, 43, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Sperandio da Silva, G.M.; Sant’Anna, C.M.; Barreiro, E.J. A novel 3D-QSAR comparative molecular field analysis (CoMFA) model of imidazole and quinazolinone functionalized p38 MAP kinase inhibitors. Bioorg. Med. Chem. 2004, 12, 3159–3166. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, G.M.S.; Lima, L.M.; Fraga, C.A.; Sant’Anna, C.M.; Barreiro, E.J. The molecular basis for coxib inhibition of p38αMAP kinase. Bioorg. Med. Chem. Lett. 2005, 15, 3506–3509. [Google Scholar] [CrossRef]
- Xiao, Z.; Varma, S.; Xiao, Y.D.; Tropsha, A. Modeling of p38 mitogen-activated protein kinase inhibitors using the Catalyst™ HypoGen and k-nearest neighbor QSAR methods. J. Mol. Graph. Model. 2004, 23, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Romeiro, N.C.; Albuquerque, M.G.; de Alencastro, R.B.; Ravi, M.; Hopfinger, A.J. Construction of 4D-QSAR models for use in the design of novel p38-MAPK inhibitors. J. Comput. Aided. Mol. Des. 2005, 19, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Edraki, N.; Hemmateenejad, B.; Miri, R.; Khoshneviszade, M. QSAR study of phenoxypyrimidine derivatives as potent inhibitors of p38 kinase using different chemometric tools. Chem. Biol. Drug Des. 2007, 70, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Li, Y.; Ren, H.; Wang, J.; Shao, L.; Zhang, S.; Li, G.; Yang, L. Insight into the structural determinants of imidazole scaffold-based derivatives as p38 MAP kinase inhibitors by computational explorations. Curr. Med. Chem. 2012, 19, 4024–4037. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.P.; Ji, Z.Q.; Zhang, H.X.; Zhang, J.W.; Wang, Y.H.; Wu, W.J. Isolation, biological evaluation and 3D-QSAR studies of insecticidal/narcotic sesquiterpene polyol esters. J. Mol. Model. 2011, 17, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, X.; Ma, M.; Zhou, W.; Wang, Y.; Yang, L. pH-Dependent channel gating in connexin26 hemichannels involves conformational changes in N-terminus. Biochim. Biophys. Acta. 2012, 1818, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, X.; Li, Y.; Wang, Y.; Yang, L. A large-scale association study for nanoparticle C60 uncovers mechanisms of nanotoxicity disrupting the native conformations of DNA/RNA. Nucleic Acids Res. 2012, 40, 7622–7632. [Google Scholar] [CrossRef] [PubMed]
- Golbraikh, A.; Shen, M.; Xiao, Z.; Xiao, Y.; Lee, K.; Tropsha, A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aided Mol. Des. 2003, 17, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Golbraikh, A.; Tropsha, A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. Mol. Divers. 2000, 5, 231–243. [Google Scholar] [CrossRef]
- Hemmateenejad, B.; Javadnia, K.; Elyasi, M. Quantitative structure-retention relationship for the Kovats retention indices of a large set of terpenes: A combined data splitting-feature selection strategy. Anal. Chim. Acta 2007, 592, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Teuvo, K. Self-organized formation of topologically correct feature maps. Biol. Cybern. 1982, 43, 59–69. [Google Scholar]
- Zupan, J.; Gasteiger, J. Neural Networks in Chemistry and Drug Design, 2nd ed.; Wiley: Weinheim, Germany, 1999. [Google Scholar]
- Juha, V.; Esa, A. Clustering of the self-organizing map. Neural Netw. IEEE Trans. 2000, 11, 586–600. [Google Scholar]
- Juha, V. SOM-based data visualization methods. Intell. Data Analys. 1999, 3, 111–126. [Google Scholar]
- Liu, H.; Gramatica, P. QSAR study of selective ligands for the thyroid hormone receptor β. Bioorg. Med. Chem. 2007, 15, 5251–5261. [Google Scholar] [CrossRef] [PubMed]
- Fortin, S.; Wei, L.; Moreau, E.; Lacroix, J.; Cote, M.F.; Petitclerc, E.; Kotra, L.P.; R, C.G. Design, synthesis, biological evaluation, and structure-activity relationships of substituted phenyl 4-(2-oxoimidazolidin-1-yl)benzenesulfonates as new tubulin inhibitors mimicking combretastatin A-4. J. Med. Chem. 2011, 54, 4559–4580. [Google Scholar] [CrossRef] [PubMed]
- Hevener, K.E.; Ball, D.M.; Buolamwini, J.K.; Lee, R.E. Quantitative structure-activity relationship studies on nitrofuranyl anti-tubercular agents. Bioorg. Med. Chem. 2008, 16, 8042–8053. [Google Scholar] [CrossRef] [PubMed]
- Juvale, D.C.; Kulkarni, V.V.; Deokar, H.S.; Wagh, N.K.; Padhye, S.B.; Kulkarni, V.M. 3D-QSAR of histone deacetylase inhibitors: Hydroxamate analogues. Org. Biomol. Chem. 2006, 4, 2858–2868. [Google Scholar] [CrossRef] [PubMed]
- Raichurkar, A.V.; Kulkarni, V.M. Understanding the antitumor activity of novel hydroxysemicarbazide derivatives as ribonucleotide reductase inhibitors using CoMFA and CoMSIA. J. Med. Chem. 2003, 46, 4419–4427. [Google Scholar] [CrossRef] [PubMed]
- Cheeseright, T.J.; Holm, M.; Lehmann, F.; Luik, S.; Gottert, M.; Melville, J.L.; Laufer, S. Novel lead structures for p38 MAP kinase via FieldScreen virtual screening. J. Med. Chem. 2009, 52, 4200–4209. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Morgan, S.L. Outlier detection in multivariate analytical chemical data. Anal. Chem. 1998, 70, 2372–2379. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.D.; Patel, M.R.; Kaushik-Basu, N.; Talele, T.T. 3D QSAR and molecular docking studies of benzimidazole derivatives as hepatitis C virus NS5B polymerase inhibitors. J. Chem. Inf. Model 2008, 48, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.P.; Hansch, C. An approach toward the problem of outliers in QSAR. Bioorg. Med. Chem. 2005, 13, 4597–4621. [Google Scholar] [CrossRef] [PubMed]
- Barken, F.M.; Gasteiger, E.L. Excitability of a penicillin-induced cortical epileptic focus. Exp. Neurol. 1980, 70, 539–547. [Google Scholar] [CrossRef]
- O’Neil, D.W.; Clark, M.V.; Lowe, J.W.; Harrington, M.S. Oral trauma in children: A hospital survey. Oral Surg. Oral Med. Oral Pathol. 1989, 68, 691–696. [Google Scholar] [CrossRef]
- Alexander, G.; Alexander, T. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar]
- Melagraki, G.; Afantitis, A. Enalos KNIME nodes: Exploring corrosion inhibition of steel in acidic medium. Chemometr. Intell. Lab. 2013, 123, 9–14. [Google Scholar] [CrossRef]
- AbdulHameed, M.D.M.; Hamza, A.; Liu, J.; Zhan, C.G. Combined 3D-QSAR modeling and molecular docking study on indolinone derivatives as inhibitors of 3-phosphoinositide-dependent protein kinase-1. J. Chem. Inf. Model. 2008, 48, 1760–1772. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha, E.F.F.; Sippl, W.; de Castro Ramalho, T.; Ceva Antunes, O.A.; de Alencastro, R.B.; Albuquerque, M.G. 3D-QSAR CoMFA/CoMSIA models based on theoretical active conformers of HOE/BAY-793 analogs derived from HIV-1 protease inhibitor complexes. Eur. J. Med. Chem. 2009, 44, 4344–4352. [Google Scholar] [CrossRef] [PubMed]
- Duda-Seiman, C.; Duda-Seiman, D.; Dragos, D.; Medeleanu, M.; Careja, V.; Putz, M.V.; Lacrama, A.-M.; Chiriac, A.; Nutiu, R.; Ciubotariu, D. Design of anti-HIV ligands by means of minimal topological difference (MTD) method. Int. J. Mol. Sci. 2006, 7, 537–555. [Google Scholar] [CrossRef]
- Duda-Seiman, D.M.; Speranţa, A.; Mancaş, S.; Careja, V.; Duda-Seiman, C.; Putz, M.V.; Ciubotariu, D. MTD-CoMSIA modelling of HMG-CoA reductase inhibitors. J. Serb. Chem. Soc. 2011, 76, 85–99. [Google Scholar] [CrossRef]
- Avram, S.; Milac, A.; Mernea, M.; Mihailescu, D.; Putz, M.V.; Buiu, C. Structure–Biological function relationship extended to mitotic arrest-deficient 2-like protein Mad2 native and mutants-new opportunity for genetic disorder control. Int. J. Mol. Sci. 2014, 15, 21381–21400. [Google Scholar] [CrossRef] [PubMed]
- Speranta, A.; Maria, M.; Dan, F.M.; Corina, D.S.; Daniel, D.S.; Mihai, V.P. Mitotic checkpoint proteins Mad1 and Mad2 structural and functional relationship with implication in genetic diseases. Curr. Comput. Aided Drug Des. 2013, 2, 1573–4099. [Google Scholar]
- Putz, M.V.; Duda-Seiman, C.; Duda-Seiman, D.M.; Putz, A.M. Turning SPECTRAL-SAR into 3D-QSAR analysis. Application on H+K+-ATPase inhibitory activity. Int. J. Chem. Model. 2008, 1, 45–62. [Google Scholar]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Sprous, D.G.; Palmer, R.K.; Swanson, J.T.; Lawless, M. QSAR in the pharmaceutical research setting: QSAR models for broad, large problems. Curr. Top Med. Chem. 2010, 10, 619–637. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Q.; Zhang, X.L.; Li, Y.; Fan, W.J.; Wang, Y.H.; Hao, M.; Zhang, S.W.; Ai, C.Z. Investigation on Quantitative Structure Activity Relationships and Pharmacophore Modeling of a Series of mGluR2 Antagonists. Int. J. Mol. Sci. 2011, 12, 5999–6023. [Google Scholar] [CrossRef] [PubMed]
- Bondavalli, F.; Botta, M.; Bruno, O.; Ciacci, A.; Corelli, F.; Fossa, P.; Lucacchini, A.; Manetti, F.; Martini, C.; Menozzi, G.; et al. Synthesis, molecular modeling studies, and pharmacological activity of selective A1 receptor antagonists. J. Med. Chem. 2002, 45, 4875–4887. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Ju, X.L.; Cheng, J.; Liu, Z.Q. 3D-QSAR studies of insecticidal anthranilic diamides as ryanodine receptor activators using CoMFA, CoMSIA and DISCOtech. Chemosphere 2010, 78, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.L.; Hao, Y.L.; Pei, J.F.; Ozoe, Y. Investigation of structural requirements for inhibitory activity at the rat and housefly picrotoxinin binding sites in ionotropic GABA receptors using DISCOtech and CoMFA. Chemosphere 2007, 69, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Floyd, J.; Gund, T.M. A comparative molecular field analysis (CoMFA) study using semiempirical, density functional, ab initio methods and pharmacophore derivation using DISCOtech on sigma 1 ligands. J. Comput. Chem. 2004, 25, 1385–1399. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Wu, M.; Ai, C.; Wang, Y. Insight into the Structural Determinants of Imidazole Scaffold-Based Derivatives as TNF-α Release Inhibitors by in Silico Explorations. Int. J. Mol. Sci. 2015, 16, 20118-20138. https://doi.org/10.3390/ijms160920118
Wang Y, Wu M, Ai C, Wang Y. Insight into the Structural Determinants of Imidazole Scaffold-Based Derivatives as TNF-α Release Inhibitors by in Silico Explorations. International Journal of Molecular Sciences. 2015; 16(9):20118-20138. https://doi.org/10.3390/ijms160920118
Chicago/Turabian StyleWang, Yuan, Mingwei Wu, Chunzhi Ai, and Yonghua Wang. 2015. "Insight into the Structural Determinants of Imidazole Scaffold-Based Derivatives as TNF-α Release Inhibitors by in Silico Explorations" International Journal of Molecular Sciences 16, no. 9: 20118-20138. https://doi.org/10.3390/ijms160920118
APA StyleWang, Y., Wu, M., Ai, C., & Wang, Y. (2015). Insight into the Structural Determinants of Imidazole Scaffold-Based Derivatives as TNF-α Release Inhibitors by in Silico Explorations. International Journal of Molecular Sciences, 16(9), 20118-20138. https://doi.org/10.3390/ijms160920118