1. Introduction
Cytochromes P450 enzymes are a family of ubiquitous heme–thiolate proteins that are involved in the metabolism of a large number of Food and Drug Administration (FDA)-approved drugs [
1]. As three of the most important isoforms within this family, CYP450 3A4, 2D6, and 2C9 catalyze nearly 50%, 25%, 20% of the therapeutic drugs currently in clinical use, respectively [
2,
3,
4]. Generally, once ingested drugs undergo biotransformation, their therapeutic efficacy always changes [
5,
6]. Therefore, metabolism mediated by these three isoforms deserve more attention during drug discovery and development.
Commonly, metabolites are determined experimentally. However, for a new chemical entity, it is common that reference compounds for its metabolites are unavailable at the time of analysis. Hence, the usage of in silico procedures to predict the metabolites can be an effective method [
7]. To predict the metabolites by in silico methods, defining the sites of metabolism (SOMs) of drugs is usually a starting point. In the current study, the chemical bond within a molecule where the metabolic reaction occurred was defined as the SOM. Therefore, identification of the SOM can provide information about which chemical bond is more susceptible to undergo biotransformation in the body. Often, this information will assist in the process of drug design and optimization. For instance, metabolites of some drugs are likely to diminish or totally lose their therapeutic efficacy; reasonable modification strategies, in ways that retain potency, can be adopted based on information about SOMs [
8,
9].
During the past decades, there have been continuous attempts in the prediction of metabolites, including ligand-based methods and structure-based methods [
10,
11,
12]. Ligand-based methods rely on the assumption that the metabolic fate of a compound is closely related to its chemical structure and characteristics [
13]. In ligand-based methods, structural information of known active and inactive compounds was used to develop prediction models. Now, there are various ligand-based models available to explore potential SOMs, including rule-based methods, pharmacophore-based methods, quantitative structure activity relationship methods, and reactivity-based
ab initio calculations [
4,
12]. Models established by ligand-based methods belong to data-driven models, because current advances in metabolism are required to generate prediction models through machine learning [
13,
14]. For this method, to certain extent, whether the model will be developed successfully directly depends on the quality of original data. However, the existing experimental data is always incomplete, because “false negatives” are unavoidable. Two reasons may account for this. On one side, some SOMs may have not been discovered based on the existing detection technology. On the other side, some SOMs have been discovered but not included in the original data [
15,
16]. After all, no databases have collected all existing metabolism data. Often, ligand-based methods are simpler and faster than structure-based methods [
7]. However, they always fail to consider the interactions between substrates and metabolic enzymes that play a pivotal role in the process of metabolism [
17]. Instead, structure-based methods simulate the interactions between substrates and metabolic enzymes successfully through molecular docking and molecular dynamics simulation [
18,
19]. Both the substrate reactivity and the shape of active sites are taken into consideration. For this method, experimental data is not a prerequisite to create the model, so there are no so-called “false negatives”. Nevertheless, there are indeed some huge challenges for SOM prediction by this method. Structure-based methods rely on the availability of 3D structures of metabolic enzymes to certain extent [
20]. However, the 3D structures of many enzymes remain unknown. Meanwhile, protein flexibility and active-site water molecules also bring about great challenges of the application of this method [
21,
22]. To accurately predict the metabolites, an integrated in silico approach that combines signal from both ligand- and structure-based methods has also been proposed [
23,
24,
25]. However, this hybrid method cannot provide a valid strategy for the problems that both ligand- and structure-based approaches suffer from.
Generally, isoform-unspecific models developed on the basis of ligand-based methods always offer information about the metabolites but fail to give information about the specific isozyme that catalyzes the metabolism [
26]. In contrast, isoform-specific models developed by ligand-based methods and models developed by structure-based methods usually only provide information about which site in a molecule is more liable to metabolism by specific CYP isozyme, but cannot determine the metabolic type of SOMs [
17,
27,
28,
29,
30]. In the current state of the art, a prediction model which can output both signal of biotransformation type of an SOM and specific isozyme catalyzing the corresponding biotransformation always draws more attention. Toward this goal, establishing prediction models based on a microsomal metabolic reaction system (MMRS) may be an effective solution. The nature of biotransformation is the break and formation of a chemical bond in the presence of metabolic enzyme. Therefore, we defined the system formed by the integration of chemical bond data and the corresponding metabolic enzyme information as MMRS. Every chemical bond can be regarded as an MMRS in combination with the corresponding enzyme. The introduction of MMRS will enable us to take the relationship between substrates and metabolic enzymes into account, independent of crystal-derived models. However, another challenge to develop a metabolism prediction model based on existing experimental data is the existence of “false negatives”. To address this issue, a negatives-screening strategy was proposed. By doing so, we attempt to minimize the risks brought by incomplete data. Actually, the complicated in vivo environment also contributes to the metabolism of drugs. Normally, the pH value in vivo ranges from 7.35 to 7.45 [
31]. To simulate the in vivo environment as much as possible, all parameters associated with substrate and enzyme were calculated under the condition of pH = 7.4.
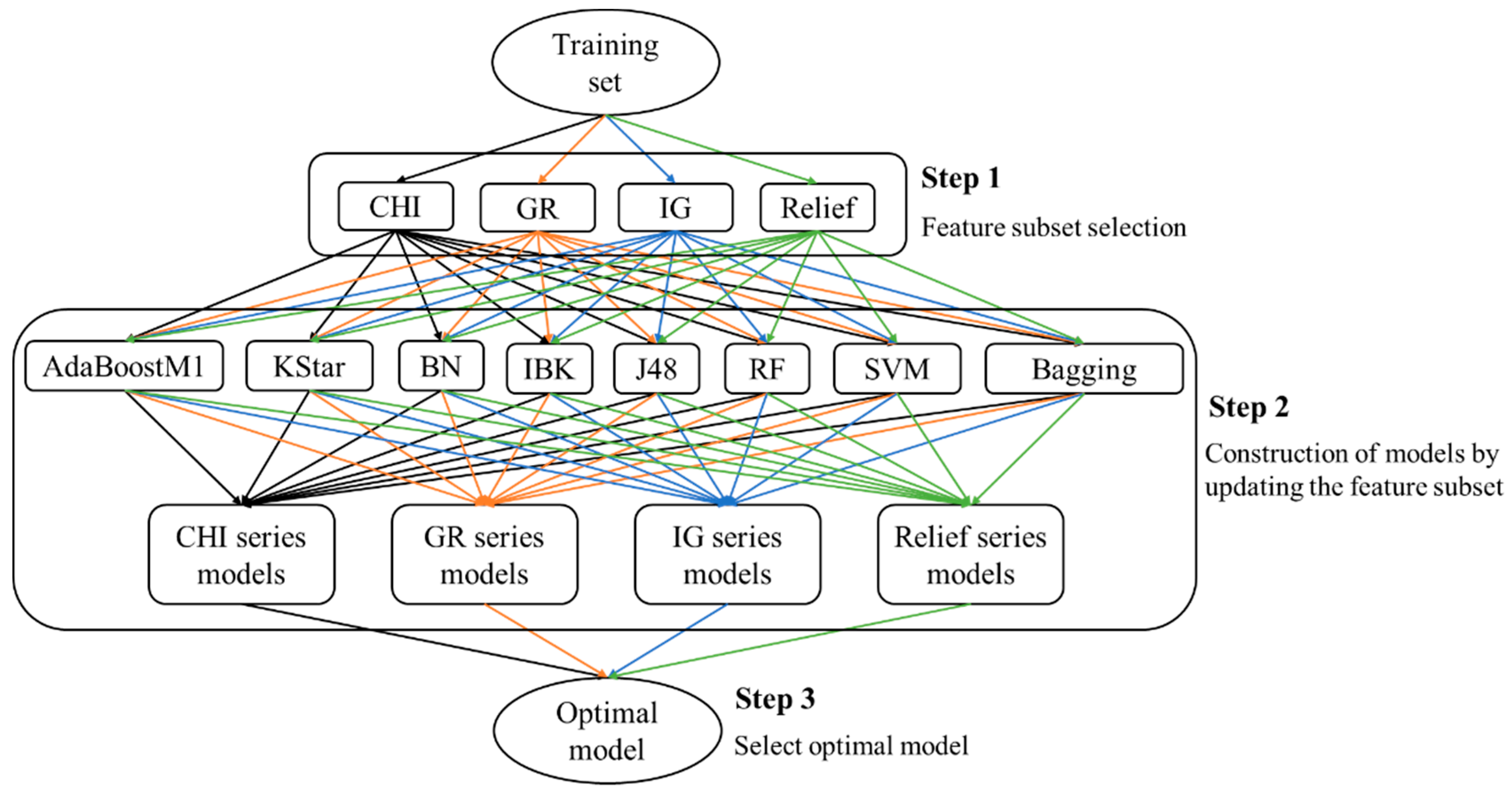
This paper presents an attempt to predict metabolism mediated by CYP3A4, 2D6, and 2C9 based on MMRS. Prediction models for the four most significant metabolic reactions were established. Firstly, all potential MMRSs were marked, and these MMRSs were categorized according to the corresponding reaction types. For each reaction type, an individual MMRS dataset was prepared. Each potential MMRS was marked as positive or unlabeled MMRS by comparing the structures of reactants and products. Then, negative MMRSs were acquired through a negatives-screening strategy. Physicochemical properties and topological properties were calculated, under the condition of pH = 7.4, to characterize the reactivity of each MMRS. These properties were used as input to train classifiers, in combination with the enzyme that catalyzes the corresponding biotransformation. To attain the optimal models, a variety of feature selection (FS) algorithms (ChiSquared (CHI), InfoGain (IG), GainRatio (GR), Relief) were used, in combination with multiple classification procedures (Kstar, Bayes (BN), K-nearest neighbours (IBK), C4.5 decision tree (J48), RandomForest (RF), Support vector machines (SVM), AdaBoostM1, Bagging). Ultimately, an optimal model for each biotransformation was attained through a comparison among the combinations of FS algorithms and classification procedures. For validation, the proposed methods were tested on their ability to recognize the reactivity of MMRSs through an independent test set. In addition, an external test was also performed based on dataset published previously. In a word, the current study aims to develop an effective approach to construction of metabolism prediction models, using only the 2D structures of the potential substrates.
2. Results
2.1. Optimal Model Selection
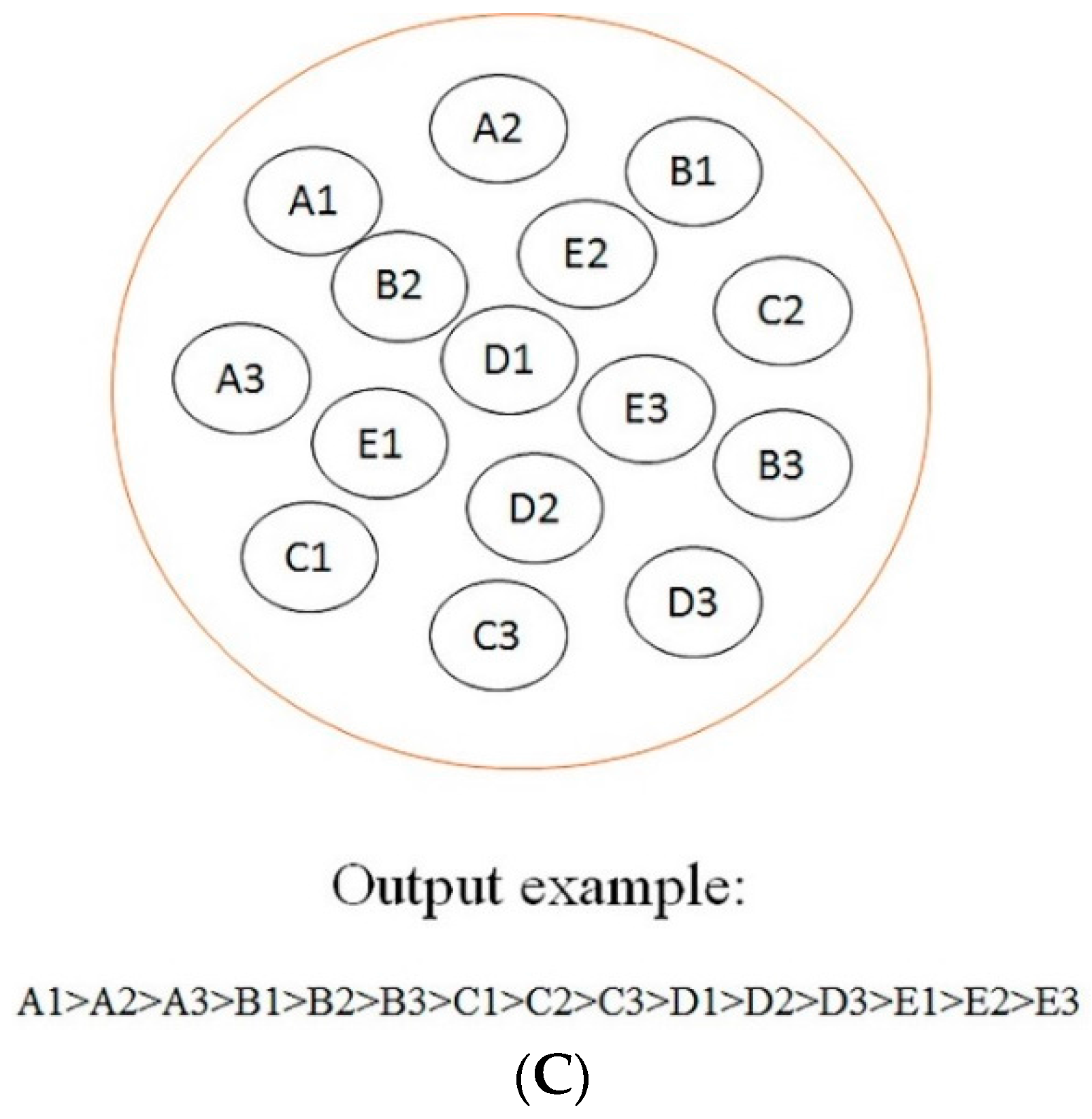
According to the optimal models screening strategy mentioned in the experimental section, four series of models were achieved for each biotransformation. Then, optimal models were acquired through a comparison among these models.
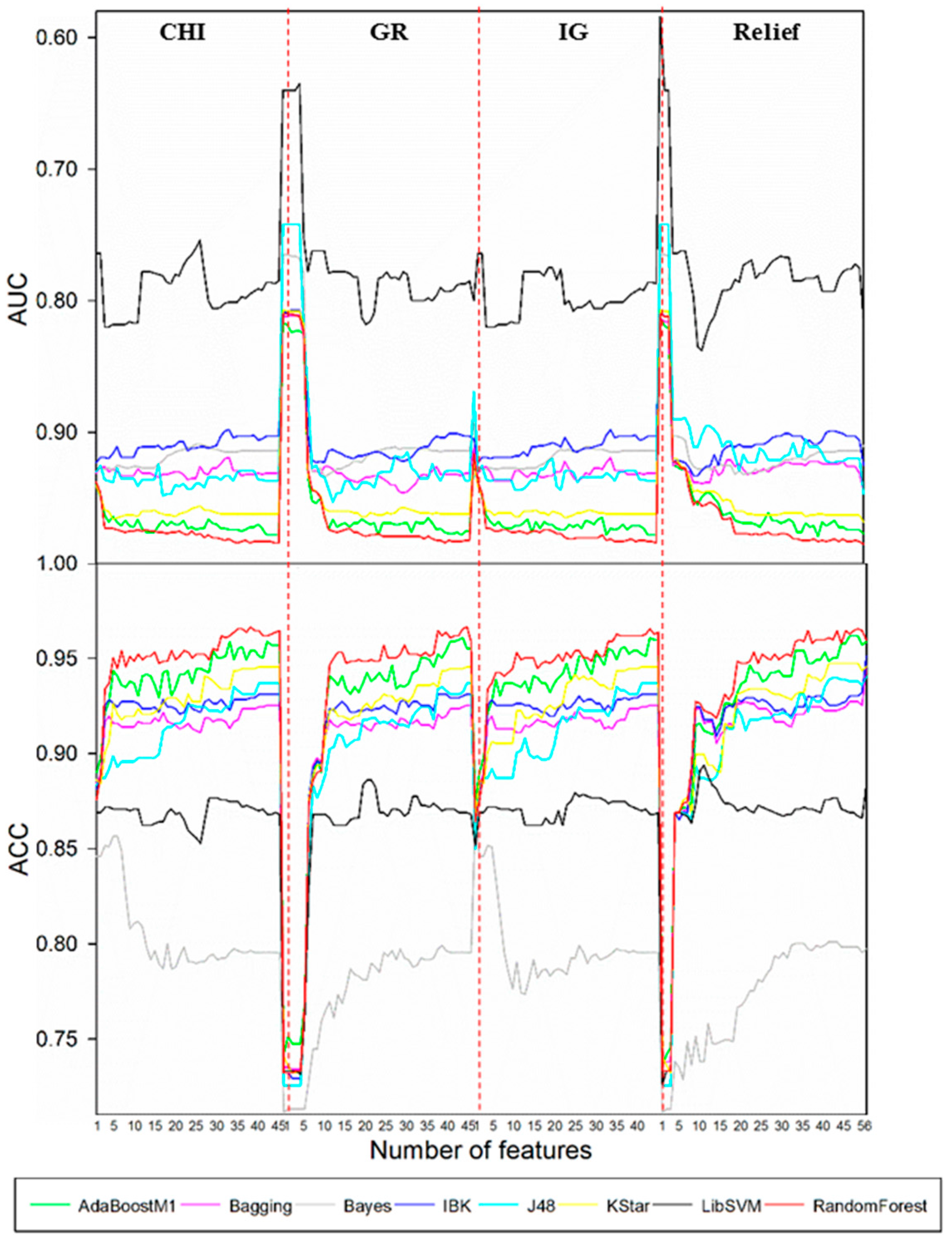
Such a selection procedure for the case of
N-dealkylation is demonstrated in
Figure 1. Results showed that the best performance was attained by the combination of CHI and RandomForest when the feature number was reduced to 39. Optimal models for other biotransformations were also obtained by the same procedure. Detailed descriptions of these models are demonstrated in
Table 1. It turned out that the optimal combinations of FS algorithms and classification procedures varied with the type of biotransformation. Both aliphatic
C-hydroxylation and
O-dealkylation attained the best performance by AdaBoostM1 classifier. However, for aromatic
C-hydroxylation and
N-dealkylation, RandomForest was more appropriate. For aliphatic
C-hydroxylation and
N-dealkylation, the most suitable FS techniques were GR and CHI, respectively. The results were in line with our assumption that there is no classification procedure or FS method that outperformed others in all applications. The optimal FS algorithms for aromatic
C-hydroxylation and
O-dealkylation were of particular interest, because they acquired the best performance when no FS was performed. Such a result indicated that in order to accurately predict the metabolites for these two biotransformations, more information may be needed than for aliphatic
C-hydroxylation and
N-dealkylation. We surmised that this circumstance may be attributed to the difference among mechanisms of these metabolic reactions. Generally,
N-dealkylation is easy to predict because of its relatively simple mechanism, but the mechanism of aromatic
C-hydroxylation is always so complicated that it is difficult to predict the metabolites. There are two alternative mechanisms for
N-dealkylation: hydrogen atom transfer and single-electron transfer. However, aromatic
C-hydroxylation proceeds via an epoxide intermediate, and the formation of the epoxide intermediate sometimes undergoes an NIH shift. In addition, direct hydroxylation through an electrophilic aromatic substitution mechanism can also occur. Such a complicated mechanism leads to a low specific SOM pattern [
32,
33,
34,
35]. The optimal feature subsets are also included in
Table 1. It was not hard to discover that the feature subset of each biotransformation covered both physicochemical properties and topological properties. Therefore, it is safe to speculate that not only physicochemical properties, but also topological properties, are associated with the reactivity of MMRS.
Through a further statistical analysis of the feature subsets, we surprisingly found that 29 out of 56 properties were observed in all cases. Such a result demonstrated that these 29 properties may play a pivotal role in the process of establishing metabolism prediction models. These properties can be further categorized as the following three categories: charge-related (1–10, 13, 17, 18, 21, 22, 25–29), adjacent atom type-related (30–33), and atom hybrid-related (45, 46, 51, 54, 55), which indicated that the reactivity of substrates was attributed to a variety of factors. In addition, we can also make the conclusion that charge-related properties make more of a contribution to the construction of metabolism prediction models than other properties. Twenty out of 29 properties mentioned above were charge-related properties, indicating the importance of charge-related properties.
2.2. Estimation of Prediction Models
To assess the overall performance of the optimal models attained above, various indices were calculated. However, it may lead to a misleading evaluation as a result of overfitting that the reliability of one model was evaluated solely by indices related to the training set. Therefore, the test set of each biotransformation was used. In the test set, the reactivity of each MMRS is currently known. They were used as input to validate the models established. Consequently, every MMRS was classified as positive or negative by our models. We believe such a test result will provide us with a more global and unbiased measure of the effectiveness of our models. Both indices associated with training set and test set were shown in
Table 2. The accuracy (ACC) of the training sets ranged from 0.928 to 0.984. The sensitivity (SE) and specificity (SP) were generally in excess of 0.953 and 0.921, respectively. For validation, the ACC of each model exceeded 0.95 while the SE varied from 0.958 to 1, and the SP of every model was >0.939. In addition, balanced accuracy (BACC) of the training set and the test set were higher than 0.937 and 0.962, respectively. All these results revealed the reliability and effectiveness of our methods of developing metabolism prediction models.
Receiver operating characteristic (ROC) analysis, another effective approach to the evaluation of the performance of a discriminative classification model, has become increasingly prevalent during the past decades. In this work, ROC analysis for both the training set and the test set was carried out. As a result, we found that every model has a significant area under curve (AUC) value, greater than 0.95, which indicated that our models have a higher reliability. The AUC of each model is also shown in
Table 2.
Note that aromatic C-hydroxylation, such a complicated biotransformation, was often hard to predict. Almost every prediction model reported has not obtained a satisfactory result. However, our model gave an ACC value of 0.928 with the AUC value = 0.950, and the independent test results were more satisfactory. Therefore, we can speculate that our methods show a superior efficiency for data fitting.
2.3. Comparison to Other Models
In this section, a comparison between our models and other models is conducted. The difference among our models, rule-based models, other ligand-based models (ligand-based models without rules-based models), and structure-based models is demonstrated in
Figure 2. As demonstrated in
Figure 2, we can find that our models focus on making a comparison among the SOMs of the same type of biotransformation, while the rule-based models tend to make a comparison among the SOMs of different biotransformations, and other ligand-based models and structure-based models aim at ranking all SOMs within a molecule according to their probabilities of undergoing biotransformations. Generally, the consequences of rules-based models, other ligand-based models, and structure-based models are reported in “molecule-scale” (where the top two ranked SOMs of a molecule containing a real SOM was thought to be a correct prediction). However, considering our models were developed from the perspective of reaction type, our model can only provide information about which SOM in a molecule is more liable to undergo certain biotransformation, but cannot provide a top-two metric. In addition, we only considered four biotransformations in the current study. However, other ligand-based models and structure-based models always failed to consider the reaction type when they predicted all possible SOMs. Therefore, there is a difference between our models and these models in terms of the reaction type. It should be noted that the model by Zheng et al. [
26]—a model which was designed to predict the SOM of six biotransformations mediated by CYP3A4, 2D6, and 2C9—was also developed from the perspective of reaction type, as we have done. However, it can only provide information about metabolites but cannot point out which isozyme catalyzes the corresponding metabolic reaction. The models developed in the current study give information about the metabolites under the catalysis of specific isozyme. Apparently, there is indeed a challenge to compare the results of Zheng et al. with ours [
26]. In summary, considering the difference between results provided by these models and ours, it is difficult to conduct a comparison between our models and others.
Why did we develop a prediction model for each biotransformation but not develop a prediction model for all four biotransformations? Firstly, during the course of the experiment, we tried to develop a prediction model for all four biotransformations, but we discovered that the result was better when we developed a prediction model for each biotransformation. In addition, such a modeling method contributes to reduction of the noise brought on other type of biotransformations. Secondly, it is easy to obtain an optimal model for each biotransformation by developing a prediction model for each biotransformation. From the results of optimal models selection, we can find that the optimal model for each biotransformation was obtained by different combinations of classification procedures and feature selection techniques, and the optimal feature subset also varied with the type of biotransformation. Apparently, such an experimental design not only improves model performance, but also contributes to obtaining the optimal model for each biotransformation.
Also, it should be emphasized that the originality of the current study does not lie in the improvement of other methods. Our research is essentially different from those researches which focus on predicting the SOMs. The main originality of our model lies in providing information about the metabolites and the enzyme which catalyzes the corresponding metabolism simultaneously. It should be noted that for CYP450 3A4, 2D6, and 2C9, currently there are no models reported that can give such detailed metabolism information.
2.4. Analysis of the Prediction Model
The originality of the methods proposed in this work was the introduction of MMRS. MMRS can represent the interaction between substrate and enzyme, to certain extent, from the integration of SOM data and enzyme information. It should be emphasized that the ligand-based methods always can only predict where a molecule may be oxidized assuming it is a substrate of certain metabolic enzyme [
36]. The proposed of MMRS made it possible to determine the metabolism of drugs and give information about metabolic enzyme simultaneously.
We concur the SMARTCyp authors’ view that false negatives (which are inevitable in the experimental data) always lead to minor signal-to-noise [
16]. We believe this is not a secret in the domain of metabolism prediction. However, almost all data-driven models lost sight of this fact. There is no doubt that the higher the quality of original data, the more reliable the models. Therefore, one advantage of the proposed methods was that a negatives-screening strategy was used to reduce the number of false negatives. By doing so, we attempt to increase the signal-to-noise ratio of original data. Another advantage of the methods was that eight classification procedures were implemented to recognize characteristic patterns in combination with four FS methods. As a result, 32 combinations of FS methods and classification procedures were generated. As we all know, there is no classifier procedure or FS method that outperforms others in all domains. Apparently, our methods are more likely to attain the optimal models than those only using one classification or FS method. The results demonstrated in
Table 1 are in line with our conjecture.
External validation is always regarded as an effective method to measure the generalization of prediction models. Therefore, the dataset collected by Sheridan et al. was used to validate these models. We believe such an external test can provide us with insight into how these models will perform in realistic problems.
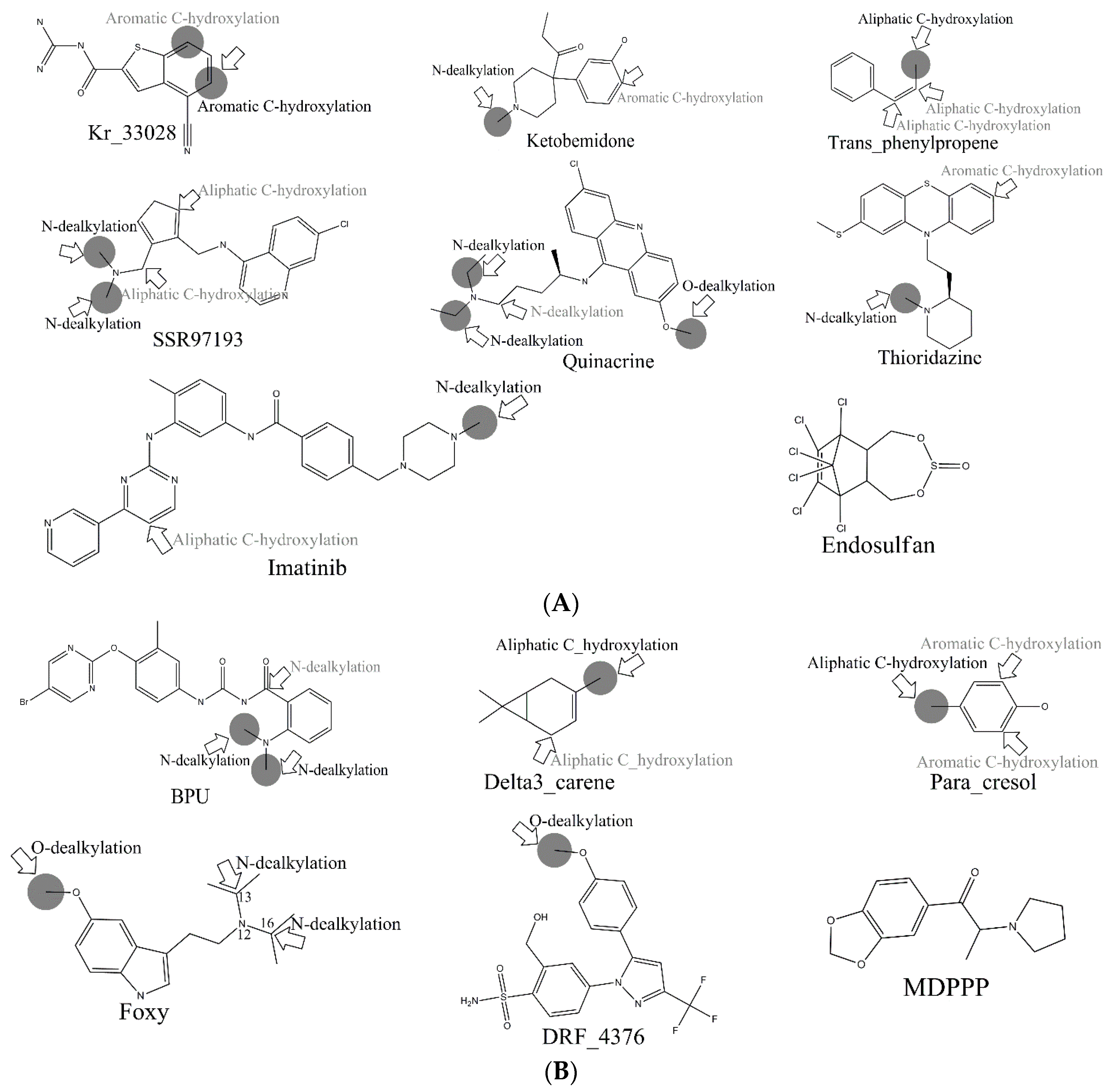
Consequently, combing our four models, 760 out of 867 MMRSs were correctly identified with an accuracy value of 0.877 for the external test set. Specifically, all the positive MMRSs were correctly identified for
N-dealkylation and
O-dealkylation. Apparently, our models gave a satisfactory performance. However, in common with all models reported, the performance of aromatic
C-hydroxylation was not so optimistic. Although we have attained the optimal model, the overall accuracy of the external set for aromatic
C-hydroxylation did not exceed 0.77. Some well-predicted and poorly-predicted compounds are shown in
Figure 3. It should be noted that Zheng et al. gave an overall accuracy value of 0.79 for the same test set based on prediction models for six metabolic reactions [
26], and neither Sheridan et al. model nor MetaSite can explain more than about 70% of the regioselectivity data [
17]. However, because of the difference of biotransformations involved between these models and ours, our models were not able to compare with them.
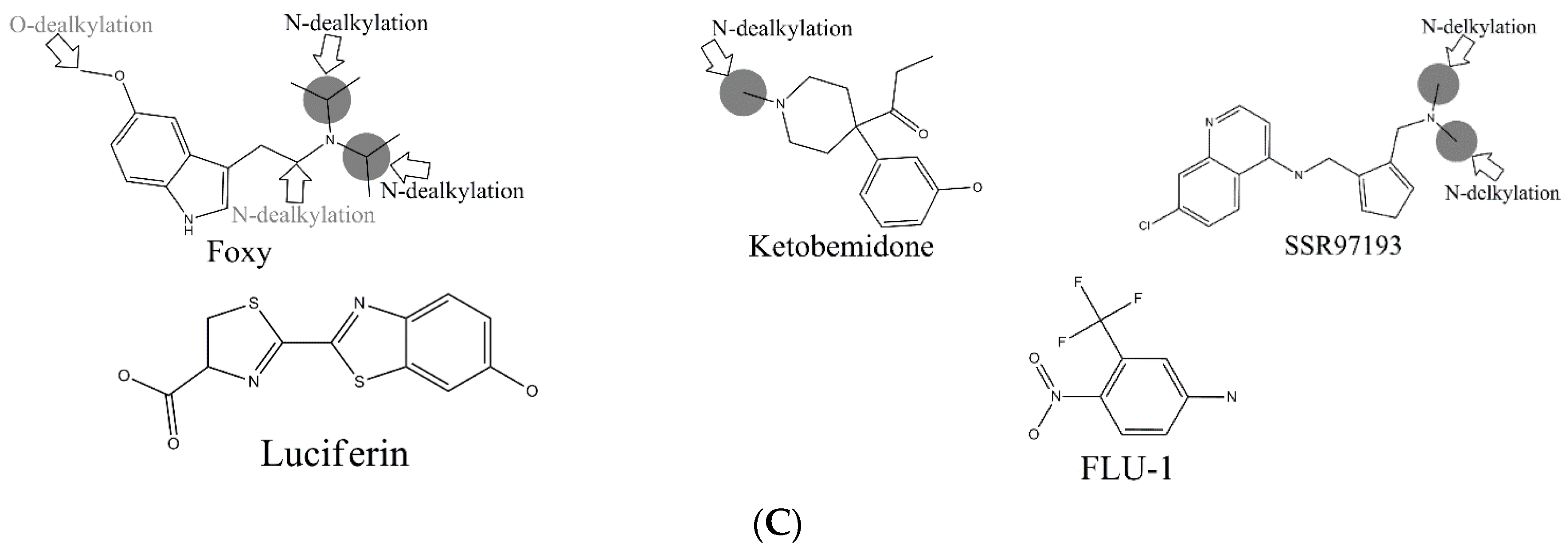
In the external test set, for the case of endosulfan, MDPPP, luciferin, and FLU-1, there are no positive SOMs within these four compounds. Fortunately, no false positives were introduced in the process of prediction. Similarly, for DRF_4376, ketobemidone, and SSR97193, our prediction results were also consistent with reports in the literature. The metabolism of Foxy, mediated by CYP450 2D6, may be more interesting. According to the records in Sheridan et al., C13-N12-CYP450 2D6 and C16-N12-CYP450 2D6 were two negative MMRSs. However, in our prediction results, they were classified as positive MMRSs. Note that this is not a fault in our models. By consulting original literature, these were confirmed as positive MMRSs which were observed experimentally [
14]. However, for other cases, the results were not so accurate: most misclassified MMRSs were attributed to aromatic
C-hydroxylation. We can be aware of this from the disappointing performance of aromatic
C-hydroxylation in
Figure 3.
Comparing the results of the external test with the independent test, we easily found that there was a difference between the performances for the model of aromatic C-hydroxylation. For the independent test, the model’s performance was fairly satisfactory. However, for the external test, the results were not so optimistic. This may be a signal that the generalization ability of this model was not so idealistic. Considering the complicated mechanism of aromatic C-hydroxylation, we believe a solution to improve the performance of this model is to consider more properties. After all, the properties considered in this work may be insufficient to completely represent such a complicated mechanism. Certainly, we also believe a more high-quality dataset also contributes to the work.
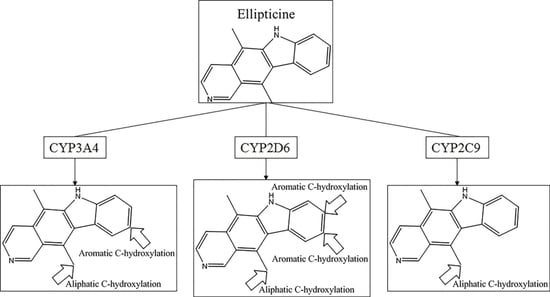
Another question that should be answered was whether the MMRS can represent the interaction between substrate and metabolic enzyme. As demonstrated in
Figure 3, it was not hard to find that the prediction results for ketobemidone, under the catalysis of CYP 3A4 and CYP 2C9, were different. Under the catalysis of CYP 3A4, an
N-dealkylation metabolite and an aromatic
C-hydroxylation metabolite were outputted. However, under the catalysis of CYP 2C9, only an
N-dealkylation metabolite was given. For SSR97193 and Foxy, we also found that the metabolites varied with metabolic enzyme. Therefore, we can speculate that MMRS can represent the interaction between substrate and enzyme to certain extent.
Metabolism prediction models for other reaction types mediated by these enzymes or other enzymes will make a significant breakthrough when sufficient data is available. Furthermore, metabolic pathway predictive model of xenobiotics will also be established based on these metabolism prediction models. However, the development of a web service for metabolism estimating and visualizing may be the first work to carry out.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



