
Validated HPAEC-PAD Method for the Determination of Fully Deacetylated Chitooligosaccharides

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
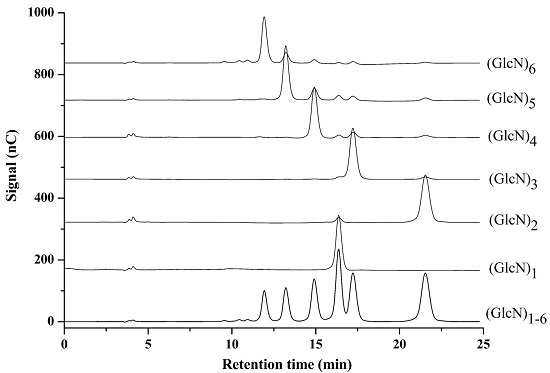
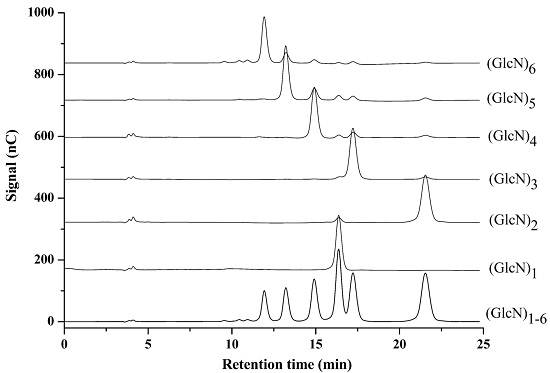
2.1. Optimization of Chromatographic Conditions
2.2. Calibration and Method Validation
2.3. Method Application
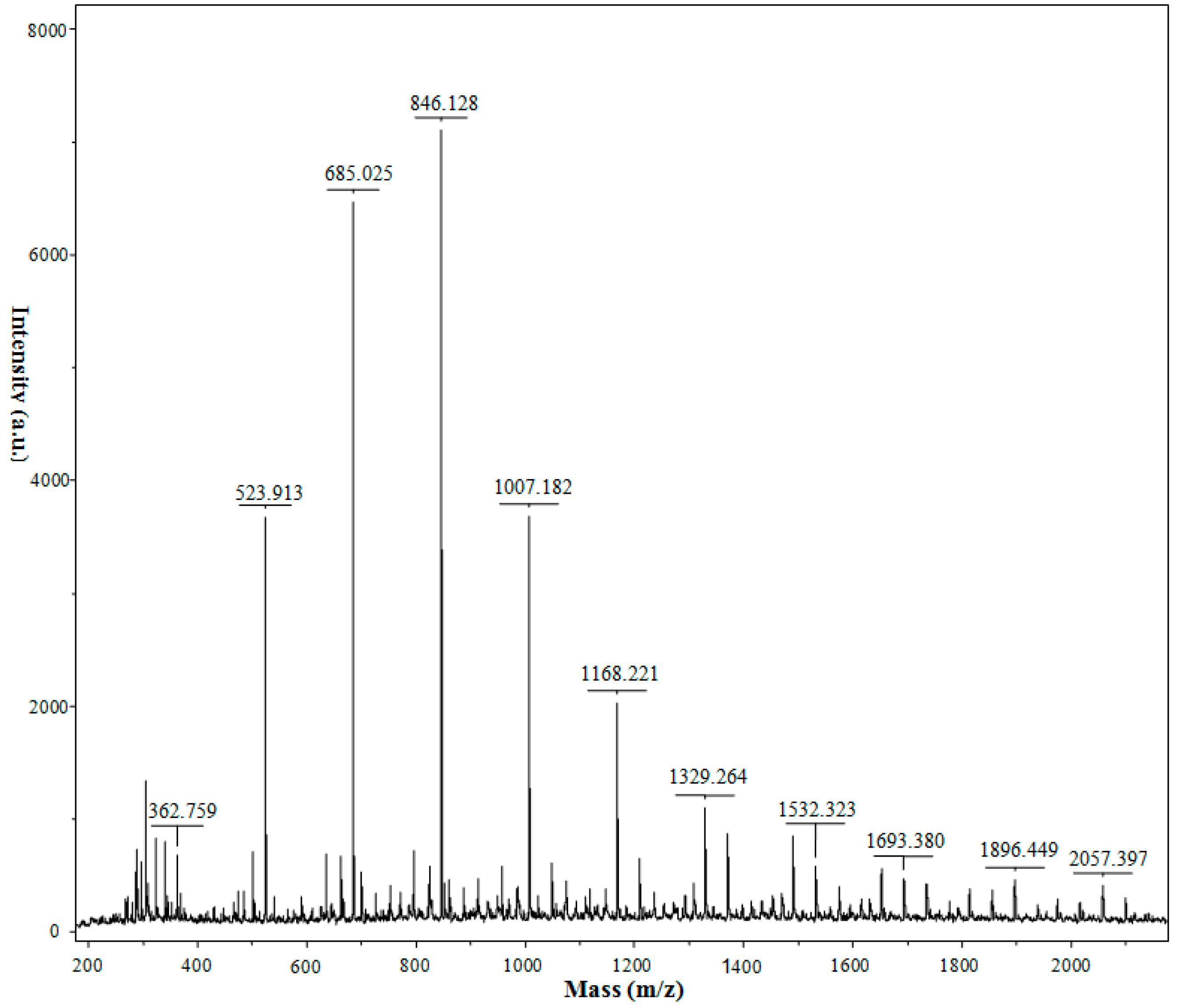
2.4. MALDI-TOF-MS Analysis
3. Experimental Section
3.1. Materials
3.2. HPAEC-PAD Analysis
3.3. Calibration
3.4. Method Validation
3.5. Hydrolysis of COS
3.6. MALDI-TOF-MS Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shahidi, F.; Arachchi, J.; Jeon, Y.J. Food applications of chitin and chitosans. Trends Food Sci. Technol. 1999, 10, 37–51. [Google Scholar] [CrossRef]
- Lee, D.X.; Xia, W.S.; Zhang, J.L. Enzymatic preparation of chitooligosaccharides by commercial lipase. Food Chem. 2008, 111, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Liu, P.; Zhang, J.; Chen, J. Biological activities of chitosan and chitooligosaccharides. Food Hydrocoll. 2011, 25, 170–179. [Google Scholar] [CrossRef]
- Li, K.; Xing, R.; Liu, S.; Li, P. Advances in preparation, analysis and biological activities of single chitooligosaccharides. Carbohydr. Polym. 2016, 139, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Aam, B.B.; Heggset, E.B.; Norberg, A.L.; Sørlie, M.; Vårum, K.M.; Eijsink, V.G.H. Production of chitooligosaccharides and their potential applications in medicine. Mar. Drugs 2010, 8, 1482–1517. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhao, X.M.; Du, Y.G. Oligochitosan: A plant diseases vaccine—A review. Carbohydr. Polym. 2010, 82, 1–8. [Google Scholar] [CrossRef]
- Park, J.K.; Chung, J.K.; Chung, M.J.; Choi, H.N.; Park, Y.I. Effects of the molecular weight and the degree of deacetylation of chitosan oligosaccharides on antitumor activity. Int. J. Mol. Sci. 2011, 12, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, J.; Hong, J.M.; Lee, S.Y.; Park, S.; Lee, J.H.; Kim, J.K. LC–MS/MS analysis of chitooligosaccharides. Carbohydr. Res. 2013, 372, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhu, X.; Li, Z.; Guo, X.; Ling, P. Application of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) in preparation of chitosan oligosaccharides (COS) with degree of polymerization (DP) 5–12 containing well-distributed acetyl groups. Int. J. Mass Spectrom. 2010, 290, 94–99. [Google Scholar] [CrossRef]
- Lang, Y.Z.; Zhao, X.; Liu, L.L.; Yu, G.L. Applications of mass spectrometry to structural analysis of marine oligosaccharides. Mar. Drugs 2014, 12, 4005–4030. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H. Enzymatic synthesis of chitooligosaccharides in organic cosolvents. Enzym. Microb. Technol. 2005, 37, 663–668. [Google Scholar] [CrossRef]
- Krokeide, I.M.; Synstad, B.; Gåseidnes, S.; Horn, S.J.; Eijsink, V.G.H.; Sørlie, M. Natural substrate assay for chitinases using high-performance liquid chromatography: A comparison with existing assays. Anal. Biochem. 2007, 363, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.Y.; Hu, Y.; Gänzle, M.G.; Lin, J.G.; Wang, C.G.; Cai, J. Preparation of chitooligosaccharides from fungal waste mycelium by recombinant chitinase. Carbohydr. Res. 2016, 430, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lopatin, S.A.; Ilyin, M.M.; Pustobaev, V.N.; Bezchetnikova, Z.A.; Varlamov, V.P.; Davankov, V.A. Mass-spectrometric analysis of N-acetylchitooligosaccharides prepared through enzymatic-hydrolysis of chitosan. Anal. Biochem. 1995, 227, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Ghauharali-van der Vlugt, K.; Bussink, A.P.; Groener, J.E.M.; Boot, R.G.; Aerts, J.M.F. Detection of chitinase activity by 2-aminobenzoic acid labeling of chito-oligosaccharides. Anal. Biochem. 2009, 384, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.J.; Souleimanov, A.; Park, R.D.; Smith, D.L. Enzymatic production of N-acetyl chitooligosaccharides by crude enzyme derived from Paenibacillus illioisensis KJA-424. Carbohydr. Polym. 2007, 67, 256–259. [Google Scholar] [CrossRef]
- Wu, H.; Yao, Z.; Bai, X.; Du, Y.; Lin, B. Anti-angiogenic activities of chitooligosaccharides. Carbohydr. Polym. 2008, 73, 105–110. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Kim, S.K. Production of chitooligosaccharides using an ultrafiltration membrane reactor and their antibacterial activity. Carbohydr. Polym. 2000, 41, 133–141. [Google Scholar] [CrossRef]
- Ji, Y.; Zhao, Y.; Zhou, X.; Weng, D.; Zhao, L. The preparation of chitosan oligomer and the components analysis. Chin. J. Modern Appl. Pharm. 2003, 2, 195–196. [Google Scholar]
- Choi, W.S.; Ahn, K.J.; Lee, D.W.; Byun, M.W.; Park, H.J. Preparation of chitosan oligomers by irradiation. Polym. Degrad. Stabil. 2002, 78, 533–538. [Google Scholar] [CrossRef]
- Hsiao, Y.C.; Lin, Y.W.; Su, C.K.; Chiang, B.H. High degree polymerized chitooligosaccharides synthesis by chitosanase in the bulk aqueous system and reversed micellar microreactors. Process Biochem. 2008, 43, 76–82. [Google Scholar] [CrossRef]
- Kittur, F.S.; Vishu Kumar, A.B.; Varadaraj, M.C.; Tharanathan, R.N. Chitooligosaccharides-Preparation with the aid of pectinase isozyme from Aspergillus niger and their antibacterial activity. Carbohydr. Res. 2005, 340, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W.; Hsiao, Y.C.; Chiang, B.H. Production of high degree polymerized chitooligosaccharides in a membrane reactor using purified chitosanase from Bacillus cereus. Food Res. Int. 2009, 42, 1355–1361. [Google Scholar] [CrossRef]
- Dong, H.; Wang, Y.; Zhao, L.; Zhou, J.; Xia, Q.; Qiu, Y. Key technologies of enzymatic preparation for DP 6–8 chitooligosaccharides. J. Food Process Eng. 2015, 38, 336–344. [Google Scholar] [CrossRef]
- Chambon, R.; Despras, G.; Brossay, A.; Vauzeilles, B.; Urban, D.; Beau, J.M.; Armand, S.; Cottaz, S.; Fort, S. Efficient chemoenzymatic synthesis of lipo-chitin oligosaccharides as plant growth promoters. Green Chem. 2015, 17, 3923–3930. [Google Scholar] [CrossRef]
- Einbu, A.; Grasdalen, H.; Varum, K.M. Kinetics of hydrolysis of chitin/chitosan oligomer in concentrated hydrochloric acid. Carbohydr. Res. 2007, 342, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Churms, S.C. Recent progress in carbohydrate separation by high-performance liquid chromatography based on hydrophilic interaction. J. Chromatogr. A 1996, 720, 75–91. [Google Scholar] [CrossRef]
- Huang, H.X.; Jin, Y.; Xue, M.Y.; Yu, L.; Fu, Q.; Ke, Y.X.; Chu, C.H.; Liang, X.M. A novel click chitooligosaccharide for hydrophilic interaction liquid chromatography. Chem. Commun. 2009, 6973–6975. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Liang, T.; Li, Z.Y.; Xu, X.Y.; Ke, Y.X.; Jin, Y.; Liang, X.M. Separation of carbohydrates using hydrophilic interaction liquid chromatography. Carbohydr. Res. 2013, 379, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Xing, R.; Liu, S.; Qin, Y.; Meng, X.; Li, P. Microwave-assisted degradation of chitosan for a possible use in inhibiting crop pathogenic fungi. Int. J. Biol. Macromol. 2012, 51, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Liu, S.; Xing, R.; Qin, Y.; Li, P. Preparation, characterization and antioxidant activity of two partially N-acetylated chitotrioses. Carbohydr. Polym. 2013, 92, 1730–1736. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Guo, Z.; Wang, C.; Yang, Y.; Liang, X.; Ding, F. Neural activity analysis of pure chito-oligomer components separated from a mixture of chitooligosaccharides. Neurosci. Lett. 2014, 581, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, M.E.; Gauthier, J.; Boucher, I.; Waldron, K.C. Capillary electrophoresis separation of a mixture of chitin and chitosan oligosaccharides derivatized using a modified fluorophore conjugation procedure. J. Sep. Sci. 2005, 28, 1390–1398. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, T.R.I.; Campa, C.; de Benedetto, G.E. Carbohydrate analysis by high-performance anion-exchange chromatography with pulsed amperometric detection: the potential is still growing. Fresenius J. Anal. Chem. 2000, 368, 739–758. [Google Scholar] [CrossRef] [PubMed]
- Lü, Y.; Yang, H.; Hu, H.; Wang, Y.; Rao, Z.; Jin, C. Mutation of Trp137 to glutamate completely removes transglycosyl activity associated with the Aspergillus fumigatus AfChiB1. Glycoconj. J. 2009, 26, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Tokuyasu, K.; Ono, H.; Hayashi, K.; Mori, Y. Reverse hydrolysis reaction of chitin deacetylase and enzymatic synthesis of β-D-GlcNAc-(1→4)-GlcN from chitobiose. Carbohydr. Res. 1999, 322, 26–31. [Google Scholar] [CrossRef]
- van Munster, J.M.; Sanders, P.; ten Kate, G.A.; Dijkhuizen, L.; van der Maarel, M.J.E.C. Kinetic characterization of Aspergillus niger chitinase CfcI using a HPAEC-PAD method for native chitin oligosaccharides. Carbohydr. Res. 2015, 407, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Mekasha, S.; Toupalová, H.; Linggadjaja, E.; Tolani, H.A.; Anděra, L.; Arntzen, M.Ø.; Vaaje-Kolstad, G.; Eijsink, V.J.H.; Agger, J.W. A novel analytical method for d-glucosamine quantification and its application in the analysis of chitosan degradation by a minimal enzyme cocktail. Carbohydr. Res. 2016, 433, 18–24. [Google Scholar] [PubMed]
- Xiong, C.; Wu, H.; Wei, P.; Pan, M.; Tuo, Y.; Kusakabe, I.; Du, Y. Potent angiogenic inhibition effects of deacetylated chitohexaose separated from chitooligosaccharides and its mechanism of action in vitro. Carbohydr. Res. 2009, 344, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, C.; Wang, Q.; Zhao, Z.K. Efficient hydrolysis of chitosan in ionic liquids. Carbohydr. Polym. 2009, 78, 685–689. [Google Scholar] [CrossRef]
- Paskach, T.J.; Licker, H.P.; Reilly, P.J.; Thielecke, K. High-performance anion-exchange chromatography of sugars and sugar alcohols on quaternary ammonium resins under alkaline conditions. Carbohydr. Res. 1991, 215, 1–14. [Google Scholar] [CrossRef]
- Rasmussen, L.E.; Meyer, A.S. Size exclusion chromatography for the quantitative profiling of the enzyme-catalyzed hydrolysis of xylo-oligosaccharides. J. Agric. Food Chem. 2010, 58, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Sørbotten, A.; Horn, S.J.; Eijsink, V.G.H.; Vårum, K.M. Degradation of chitosans with chitinase B from Serratia marcescens-Production of chito-oligosaccharides and insight into enzyme processivity. FEBS J. 2005, 272, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Borromei, C.; Cavazza, A.; Merusi, C.; Corradini, C. Characterization and quantitation of short-chain fructooligosaccharides and inulooligosaccharides in fermented milks by high-performance anion-exchange chromatography with pulsed amperometric detection. J. Sep. Sci. 2009, 32, 3635–3642. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.; Andersson, R.; Åman, P. Quantitative analysis of amylopectin unit chains by means of highperformance anion-exchange chromatography with pulsed amperometric detection. J. Chromatogr. A 1998, 800, 199–206. [Google Scholar] [CrossRef]
- SANCO/3030/99 rev.4, 11/07/00, Technical Material and Preparations: Guidance for generating and reporting methods of analysis in support of pre-and post-registration data requirements for Annex II (part A, Section 4) and Annex III (part A, Section 5) of Directive 91/414. Available online: http://ec.europa.eu/food/fs/sfp/ph_ps/pro/wrkdoc/wrkdoc13_en.pdf (accessed on 9 October 2016).
- Jank, L.; Hoff, R.B.; da Costa, F.J.; Pizzolato, P.M. Simultaneous determination of eight antibiotics from distinct classes in surface and wastewater samples by solid-phase extraction and high performance liquid chromatography–electrospray ionisation mass spectrometry. Int. J. Environ. Anal. Chem. 2014, 94, 1013–1037. [Google Scholar] [CrossRef]
- Martins, M.T.; Barreto, F.; Hoff, R.B.; Arsand, J.B.; Motta, T.M.C.; Schapoval, E.E.S. Multiclass and multi-residue determination of antibiotics in bovine milk by liquid chromatographyetandem mass spectrometry: Combining efficiency of milk control and simplicity of routine analysis. Int. Dairy J. 2016, 59, 44–51. [Google Scholar] [CrossRef]
- Fraselle, S.; Derop, V.; Degroodt, J.-M.; van Loco, J. Validation of a method for the detection and confirmation of nitroimidazoles and the corresponding hydroxy metabolites in pig plasma by high performance liquid chromatography–tandem mass spectrometry. Anal. Chim. Acta 2007, 586, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, J.L.; Bu, F.; Xia, W.S. Determination of chitosan with a modified acid hydrolysis and HPLC method. Carbohydr. Res. 2013, 366, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Field, M.; Papac, D.; Jones, A. The use of high-performance anion-exchange chromatography and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry to monitor and identify oligosaccharide degradation. Anal. Biochem. 1996, 239, 92–98. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Linear Range (mg/L) | Calibration Curve a | R2 | LOD (mg/L) | LOQ (mg/L) |
|---|---|---|---|---|---|
| (GlcN)1 | 0.2–11.5 | y = 14.455x + 0.5347 | 0.9994 | 0.003 | 0.009 |
| (GlcN)2 | 0.2–9.8 | y = 7.6363x + 1.0974 | 0.9984 | 0.008 | 0.027 |
| (GlcN)3 | 0.2–10.1 | y = 7.0084x + 0.9652 | 0.9988 | 0.008 | 0.027 |
| (GlcN)4 | 0.2–10.6 | y = 5.4722x + 0.6928 | 0.9979 | 0.011 | 0.035 |
| (GlcN)5 | 0.2–10.6 | y = 4.5079x + 0.0593 | 0.9995 | 0.013 | 0.045 |
| (GlcN)6 | 0.2–9.9 | y = 3.1615x + 0.3702 | 0.9992 | 0.016 | 0.054 |
| Analyte | Repeatability (n = 7) | Intermediate Precision (n = 9) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Peak Area | Retention Time | Peak Area | Retention Time | |||||||||
| C1 | C2 | C3 | C1 | C2 | C3 | C1 | C2 | C3 | C1 | C2 | C3 | |
| (GlcN)1 | 2.77 | 2.71 | 2.06 | 0.11 | 0.40 | 0.28 | 2.51 | 2.67 | 2.29 | 0.41 | 0.59 | 0.67 |
| (GlcN)2 | 4.09 | 2.38 | 4.16 | 0.12 | 0.53 | 0.34 | 4.17 | 3.63 | 4.27 | 0.54 | 0.54 | 0.90 |
| (GlcN)3 | 3.16 | 2.22 | 2.91 | 0.14 | 0.54 | 0.35 | 2.92 | 2.79 | 3.17 | 0.59 | 0.58 | 0.92 |
| (GlcN)4 | 2.66 | 0.99 | 2.95 | 0.11 | 0.57 | 0.36 | 2.38 | 2.65 | 2.66 | 0.58 | 0.58 | 0.97 |
| (GlcN)5 | 2.96 | 0.90 | 3.24 | 0.13 | 0.58 | 0.38 | 2.95 | 2.66 | 4.04 | 0.60 | 0.60 | 0.99 |
| (GlcN)6 | 4.67 | 1.54 | 3.95 | 0.12 | 0.56 | 0.40 | 4.71 | 1.74 | 6.63 | 0.66 | 0.66 | 1.02 |
| Analyte | Content (%, m/m) a | Mean (%) | %RSD | %RSDr | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||||
| (GlcN)1 | 0.16 | 0.16 | 0.17 | 0.16 | 0.16 | 0.15 | 0.16 | 0.16 | 3.19 | 3.53 |
| (GlcN)2 | 2.52 | 2.44 | 2.52 | 2.59 | 2.53 | 2.55 | 2.50 | 2.52 | 1.83 | 2.33 |
| (GlcN)3 | 5.44 | 5.55 | 5.66 | 5.55 | 5.56 | 5.64 | 5.74 | 5.59 | 1.70 | 2.07 |
| (GlcN)4 | 9.73 | 9.86 | 9.55 | 9.67 | 10.09 | 9.78 | 9.67 | 9.76 | 1.80 | 1.90 |
| (GlcN)5 | 11.42 | 11.08 | 11.21 | 11.26 | 10.93 | 11.07 | 10.92 | 11.13 | 1.64 | 1.86 |
| (GlcN)6 | 8.36 | 8.47 | 8.54 | 8.25 | 8.37 | 8.48 | 8.62 | 8.44 | 1.49 | 1.94 |
| Analyte | Recovery (%) | ||
|---|---|---|---|
| Spiked C1 | Spiked C2 | Spiked C3 | |
| (GlcN)1 | 91.16 ± 3.22 | 103.74 ± 0.79 | 95.47 ± 2.73 |
| (GlcN)2 | 97.59 ± 0.96 | 97.81 ± 2.93 | 95.98 ± 1.53 |
| (GlcN)3 | 94.01 ± 1.08 | 95.93 ± 0.13 | 99.67 ± 3.26 |
| (GlcN)4 | 95.08 ± 0.60 | 98.97 ± 2.21 | 98.42 ± 2.35 |
| (GlcN)5 | 94.94 ± 0.63 | 97.94 ± 3.02 | 103.94 ± 2.16 |
| (GlcN)6 | 97.11 ± 0.54 | 94.17 ± 0.45 | 97.42 ± 2.46 |
| Chitooligosaccharides | m/z [M + Na]+ | DP |
|---|---|---|
| (GlcN)2 | 363 | 2 |
| (GlcN)3 | 524 | 3 |
| (GlcN)4 | 685 | 4 |
| (GlcN)5 | 846 | 5 |
| (GlcN)6 | 1007 | 6 |
| (GlcN)5(GlcNAc)1 | 1049 | 6 |
| (GlcN)7 | 1168 | 7 |
| (GlcN)6(GlcNAc)1 | 1210 | 7 |
| (GlcN)8 | 1329 | 8 |
| (GlcN)7(GlcNAc)1 | 1371 | 8 |
| (GlcN)9 | 1490 | 9 |
| (GlcN)8(GlcNAc)1 | 1532 | 9 |
| (GlcN)7(GlcNAc)2 | 1574 | 9 |
| (GlcN)10 | 1651 | 10 |
| (GlcN)9(GlcNAc)1 | 1693 | 10 |
| (GlcN)8(GlcNAc)2 | 1735 | 10 |
| (GlcN)7(GlcNAc)3 | 1777 | 10 |
| (GlcN)11 | 1812 | 11 |
| (GlcN)10(GlcNAc)1 | 1854 | 11 |
| (GlcN)9(GlcNAc)2 | 1896 | 11 |
| (GlcN)8(GlcNAc)3 | 1938 | 11 |
| (GlcN)12 | 1973 | 12 |
| (GlcN)11(GlcNAc)1 | 2015 | 12 |
| (GlcN)10(GlcNAc)2 | 2057 | 12 |
| (GlcN)9(GlcNAc)3 | 2099 | 12 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, L.; Wu, J.; Li, X.; Zheng, L.; Wu, M.; Liu, P.; Huang, Q. Validated HPAEC-PAD Method for the Determination of Fully Deacetylated Chitooligosaccharides. Int. J. Mol. Sci. 2016, 17, 1699. https://doi.org/10.3390/ijms17101699
Cao L, Wu J, Li X, Zheng L, Wu M, Liu P, Huang Q. Validated HPAEC-PAD Method for the Determination of Fully Deacetylated Chitooligosaccharides. International Journal of Molecular Sciences. 2016; 17(10):1699. https://doi.org/10.3390/ijms17101699
Chicago/Turabian StyleCao, Lidong, Jinlong Wu, Xiuhuan Li, Li Zheng, Miaomiao Wu, Pingping Liu, and Qiliang Huang. 2016. "Validated HPAEC-PAD Method for the Determination of Fully Deacetylated Chitooligosaccharides" International Journal of Molecular Sciences 17, no. 10: 1699. https://doi.org/10.3390/ijms17101699
APA StyleCao, L., Wu, J., Li, X., Zheng, L., Wu, M., Liu, P., & Huang, Q. (2016). Validated HPAEC-PAD Method for the Determination of Fully Deacetylated Chitooligosaccharides. International Journal of Molecular Sciences, 17(10), 1699. https://doi.org/10.3390/ijms17101699