
Learning the Relationship between the Primary Structure of HIV Envelope Glycoproteins and Neutralization Activity of Particular Antibodies by Using Artificial Neural Networks




Abstract
:
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Collecting Data
4.2. Creating the Network
4.3. Configure the Network
4.4. Initializing the Network
4.5. Training the Network
4.6. Validating the Network
4.7. Utilizing the Network
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Julien, J.-P.; Cupo, A.; Sok, D.; Stanfield, R.L.; Lyumkis, D.; Deller, M.C.; Klasse, P.-J.; Burton, D.R.; Sanders, R.W.; Moore, J.P.; et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 2013, 342, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Foley, B.; Apetrei, C.; Mizrachi, I.; Rambaut, A.; Korber, B.; Kuiken, C.; Leitner, T.; Hahn, B.; Mullins, J.; Wolinsky, S.; et al. HIV Sequence Compendium 2012; Los Alamos National Laboratory, Theoretical Biology and Biophysics: Los Alamos, NM, USA, 2012.
- Pancera, M.; Majeed, S.; Ban, Y.-E.A.; Chen, L.; Huang, C.; Kong, L.; Do Kwon, Y.; Stuckey, J.; Zhou, T.; Robinson, J.E.; et al. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc. Natl. Acad. Sci. USA 2010, 107, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Merk, A.; Subramaniam, S. HIV-1 envelope glycoprotein structure. Curr. Opin. Struct. Biol. 2013, 23, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Tang, M.; Zhang, M.-Y.; Majeed, S.; Montabana, E.; Stanfield, R.L.; Dimitrov, D.S.; Korber, B.; Sodroski, J.; Wilson, I.A.; et al. Structure of a V3-containing HIV-1 gp120 core. Science 2005, 310, 1025–1028. [Google Scholar] [CrossRef] [PubMed]
- Heider, D.; Dybowski, J.N.; Wilms, C.; Hoffmann, D. A simple structure-based model for the prediction of HIV-1 co-receptor tropism. BioData Min. 2014, 7, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Cashin, K.; Sterjovski, J.; Harvey, K.L.; Ramsland, P.A.; Churchill, M.J.; Gorry, P.R. Covariance of charged amino acids at positions 322 and 440 of HIV-1 Env contributes to coreceptor specificity of subtype B viruses, and can be used to improve the performance of V3 sequence-based coreceptor usage prediction algorithms. PLoS ONE 2014, 9, e109771. [Google Scholar] [CrossRef] [PubMed]
- Shityakov, S.; Dandekar, T. Lead expansion and virtual screening of Indinavir derivate HIV-1 protease inhibitors using pharmacophoric—Shape similarity scoring function. Bioinformation 2010, 4, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Harte, W.E.; Swaminathan, S.; Beveridge, D.L. Molecular dynamics of HIV-1 protease. Proteins 1992, 13, 175–194. [Google Scholar] [CrossRef] [PubMed]
- Shityakov, S.; Rethwilm, A.; Dandekar, T. Structural and docking analysis of HIV-1 integrase and Transportin-SR2 interaction: Is this a more general and specific route for retroviral nuclear import and its regulation? Online J. Bioinform. 2010, 11, 19–33. [Google Scholar]
- Kwong, P.D.; Mascola, J.R. Human antibodies that neutralize HIV-1: Identification, structures, and B cell ontogenies. Immunity 2012, 37, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Frey, G.; Peng, H.; Rits-Volloch, S.; Garrity, J.; Seaman, M.S.; Chen, B. Mechanism of HIV-1 neutralization by antibodies targeting a membrane-proximal region of gp41. J. Virol. 2014, 88, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Hessell, A.J.; Rakasz, E.G.; Poignard, P.; Hangartner, L.; Landucci, G.; Forthal, D.N.; Koff, W.C.; Watkins, D.I.; Burton, D.R. Broadly neutralizing human anti-HIV antibody 2G12 is effective in protection against mucosal SHIV challenge even at low serum neutralizing titers. PLoS Pathog. 2009, 5, e1000433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Wang, P. Passive immunization against HIV/AIDS by antibody gene transfer. Viruses 2014, 6, 428–447. [Google Scholar] [CrossRef] [PubMed]
- Kepler, T.B.; Liao, H.-X.; Alam, S.M.; Bhaskarabhatla, R.; Zhang, R.; Yandava, C.; Stewart, S.; Anasti, K.; Kelsoe, G.; Parks, R.; et al. Immunoglobulin gene insertions and deletions in the affinity maturation of HIV-1 broadly reactive neutralizing antibodies. Cell Host Microbe 2014, 16, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yang, Z.-Y.; Li, Y.; Hogerkorp, C.-M.; Schief, W.R.; Seaman, M.S.; Zhou, T.; Schmidt, S.D.; Wu, L.; Xu, L.; et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 2010, 329, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Scheid, J.F.; Mouquet, H.; Ueberheide, B.; Diskin, R.; Klein, F.; Oliveira, T.Y.K.; Pietzsch, J.; Fenyo, D.; Abadir, A.; Velinzon, K.; et al. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science 2011, 333, 1633–1637. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhou, T.; Zhu, J.; Zhang, B.; Georgiev, I.; Wang, C.; Chen, X.; Longo, N.S.; Louder, M.; McKee, K.; et al. Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science 2011, 333, 1593–1602. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.M.; Huber, M.; Doores, K.J.; Falkowska, E.; Pejchal, R.; Julien, J.-P.; Wang, S.-K.; Ramos, A.; Chan-Hui, P.-Y.; Moyle, M.; et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 2011, 477, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Bouvin-Pley, M.; Morgand, M.; Meyer, L.; Goujard, C.; Moreau, A.; Mouquet, H.; Nussenzweig, M.; Pace, C.; Ho, D.; Bjorkman, P.J.; et al. Drift of the HIV-1 envelope glycoprotein gp120 toward increased neutralization resistance over the course of the epidemic: A comprehensive study using the most potent and broadly neutralizing monoclonal antibodies. J. Virol. 2014, 88, 13910–13917. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.S.; Leaman, D.P.; Zwick, M.B. Antibody to gp41 MPER Alters Functional Properties of HIV-1 Env without Complete Neutralization. PLoS Pathog. 2014, 10, e1004271. [Google Scholar] [CrossRef] [PubMed]
- Bradley, T.; Trama, A.; Tumba, N.; Gray, E.; Lu, X.; Madani, N.; Jahanbakhsh, F.; Eaton, A.; Xia, S.-M.; Parks, R.; et al. Amino acid changes in the HIV-1 gp41 membrane proximal region control virus neutralization sensitivity. EBioMedicine 2016, 16, 30402–30409. [Google Scholar] [CrossRef] [PubMed]
- Ofek, G.; Zirkle, B.; Yang, Y.; Zhu, Z.; McKee, K.; Zhang, B.; Chuang, G.-Y.; Georgiev, I.S.; O’Dell, S.; Doria-Rose, N.; et al. Structural basis for HIV-1 neutralization by 2F5-like antibodies m66 and m66.6. J. Virol. 2014, 88, 2426–2441. [Google Scholar] [CrossRef] [PubMed]
- Shmelkov, E.; Grigoryan, A.; Krachmarov, C.; Abagyan, R.; Cardozo, T.J. Sequence conserved and antibody accessible sites in the V1V2 domain of HIV-1 gp120 envelope protein. AIDS Res. Hum. Retrovir. 2014, 30, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Bouvin-Pley, M.; Morgand, M.; Moreau, A.; Jestin, P.; Simonnet, C.; Tran, L.; Goujard, C.; Meyer, L.; Barin, F.; Braibant, M. Evidence for a continuous drift of the HIV-1 species towards higher resistance to neutralizing antibodies over the course of the epidemic. PLoS Pathog. 2013, 9, e1003477. [Google Scholar] [CrossRef] [PubMed]
- Frey, G.; Peng, H.; Rits-Volloch, S.; Morelli, M.; Cheng, Y.; Chen, B. A fusion-intermediate state of HIV-1 gp41 targeted by broadly neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2008, 105, 3739–3744. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, B.K.; Walker, L.M.; Guenaga, J.F.; Ghobbeh, A.; Poignard, P.; Burton, D.R.; Wyatt, R.T. Direct antibody access to the HIV-1 membrane-proximal external region positively correlates with neutralization sensitivity. J. Virol. 2011, 85, 8217–8226. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Scharf, L.; Horwitz, J.; Klein, F.; Nussenzweig, M.C.; Bjorkman, P.J. Computational analysis of anti-HIV-1 antibody neutralization panel data to identify potential functional epitope residues. Proc. Natl. Acad. Sci. USA 2013, 110, 10598–10603. [Google Scholar] [CrossRef] [PubMed]
- Montefiori, D.C. Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. Curr. Protoc. Immunol. 2005, 12. [Google Scholar] [CrossRef]
- Kohavi, R.; Provost, F. Glossary of Terms. Mach. Learn. 1998, 30, 271–274. [Google Scholar]
- Mitchell, T.M. Machine Learning; McGraw-Hill, Inc.: New York, NY, USA, 1997. [Google Scholar]
- Al-Gharabli, S.I.; Al-Agtash, S.; Rawashdeh, N.A.; Barqawi, K.R. Artificial neural networks for dihedral angles prediction in enzyme loops: A novel approach. Int. J. Bioinform. Res. Appl. 2015, 11, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Ashtawy, H.M.; Mahapatra, N.R. BgN-Score and BsN-Score: Bagging and boosting based ensemble neural networks scoring functions for accurate binding affinity prediction of protein-ligand complexes. BMC Bioinform. 2015, 16, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Kourou, K.; Exarchos, T.P.; Exarchos, K.P.; Karamouzis, M.V.; Fotiadis, D.I. Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J. 2015, 13, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Wrede, P. Artificial neural networks for computer-based molecular design. Prog. Biophys. Mol. Biol. 1998, 70, 175–222. [Google Scholar] [CrossRef]
- Douali, L.; Villemin, D.; Zyad, A.; Cherqaoui, D. Artificial neural networks: Non-linear QSAR studies of HEPT derivatives as HIV-1 reverse transcriptase inhibitors. Mol. Divers. 2004, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.A. Neural networks as robust tools in drug lead discovery and development. Mol. Biotechnol. 2004, 27, 139–168. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. NNScore: A neural-network-based scoring function for the characterization of protein-ligand complexes. J. Chem. Inf. Model. 2010, 50, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.V. Chi-Sang Poon Linear independence of internal representations in multilayer perceptrons. IEEE Trans. Neural Networks 1999, 10, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, D.W. An algorithm for least-squares estimation of nonlinear parameters. J. Soc. Ind. Appl. Math. 1963, 11, 431–441. [Google Scholar] [CrossRef]
- Hagan, M.T.; Menhaj, M.B. Training feedforward networks with the Marquardt algorithm. IEEE Trans. Neural Networks 1994, 5, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Milac, A.-L.; Avram, S.; Petrescu, A.-J. Evaluation of a neural networks QSAR method based on ligand representation using substituent descriptors: Application to HIV-1 protease inhibitors. J. Mol. Graph. Model. 2006, 25, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Calborean, O.; Mernea, M.; Avram, S.; Mihailescu, D.F. Pharmacological descriptors related to the binding of Gp120 to CD4 corresponding to 60 representative HIV-1 strains. J. Enzym. Inhib. Med. Chem. 2013, 28, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Wold, S.; Sjostrom, M.; Eriksson, L. PLS-regression: A basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar]
- Qian, N.; Sejnowski, T.J. Predicting the secondary structure of globular proteins using neural network models. J. Mol. Biol. 1988, 202, 865–884. [Google Scholar] [CrossRef]
- Heider, D.; Hoffmann, D. Interpol: An R package for preprocessing of protein sequences. BioData Min. 2011, 4, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Kuiken, C.; Korber, B.; Shafer, R.W. HIV sequence databases. AIDS Rev. 2003, 5, 52–61. [Google Scholar] [PubMed]
- Diskin, R.; Scheid, J.F.; Marcovecchio, P.M.; West, A.P.; Klein, F.; Gao, H.; Gnanapragasam, P.N.P.; Abadir, A.; Seaman, M.S.; Nussenzweig, M.C.; et al. Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science 2011, 334, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Diskin, R.; Klein, F.; Horwitz, J.A.; Halper-Stromberg, A.; Sather, D.N.; Marcovecchio, P.M.; Lee, T.; West, A.P.; Gao, H.; Seaman, M.S.; et al. Restricting HIV-1 pathways for escape using rationally designed anti-HIV-1 antibodies. J. Exp. Med. 2013, 210, 1235–1249. [Google Scholar] [CrossRef] [PubMed]
- Mouquet, H.; Scharf, L.; Euler, Z.; Liu, Y.; Eden, C.; Scheid, J.F.; Halper-Stromberg, A.; Gnanapragasam, P.N.P.; Spencer, D.I.R.; Seaman, M.S.; et al. Complex-type N-glycan recognition by potent broadly neutralizing HIV antibodies. Proc. Natl. Acad. Sci. USA 2012, 109, 3268–3277. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ofek, G.; Laub, L.; Louder, M.K.; Doria-Rose, N.A.; Longo, N.S.; Imamichi, H.; Bailer, R.T.; Chakrabarti, B.; Sharma, S.K.; et al. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 2012, 491, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Doria-Rose, N.A.; Louder, M.K.; Yang, Z.; O’Dell, S.; Nason, M.; Schmidt, S.D.; McKee, K.; Seaman, M.S.; Bailer, R.T.; Mascola, J.R. HIV-1 neutralization coverage is improved by combining monoclonal antibodies that target independent epitopes. J. Virol. 2012, 86, 3393–3397. [Google Scholar] [CrossRef] [PubMed]
- Buiu, C. Neutralization Data and Aligned ENV Sequences for Predicting Antibody Affinities Using Artificial Neural Networks. Mendeley Data. Available online: http://dx.doi.org/10.17632/bhcjwtwjh4.1 (accessed on 8 October 2016).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
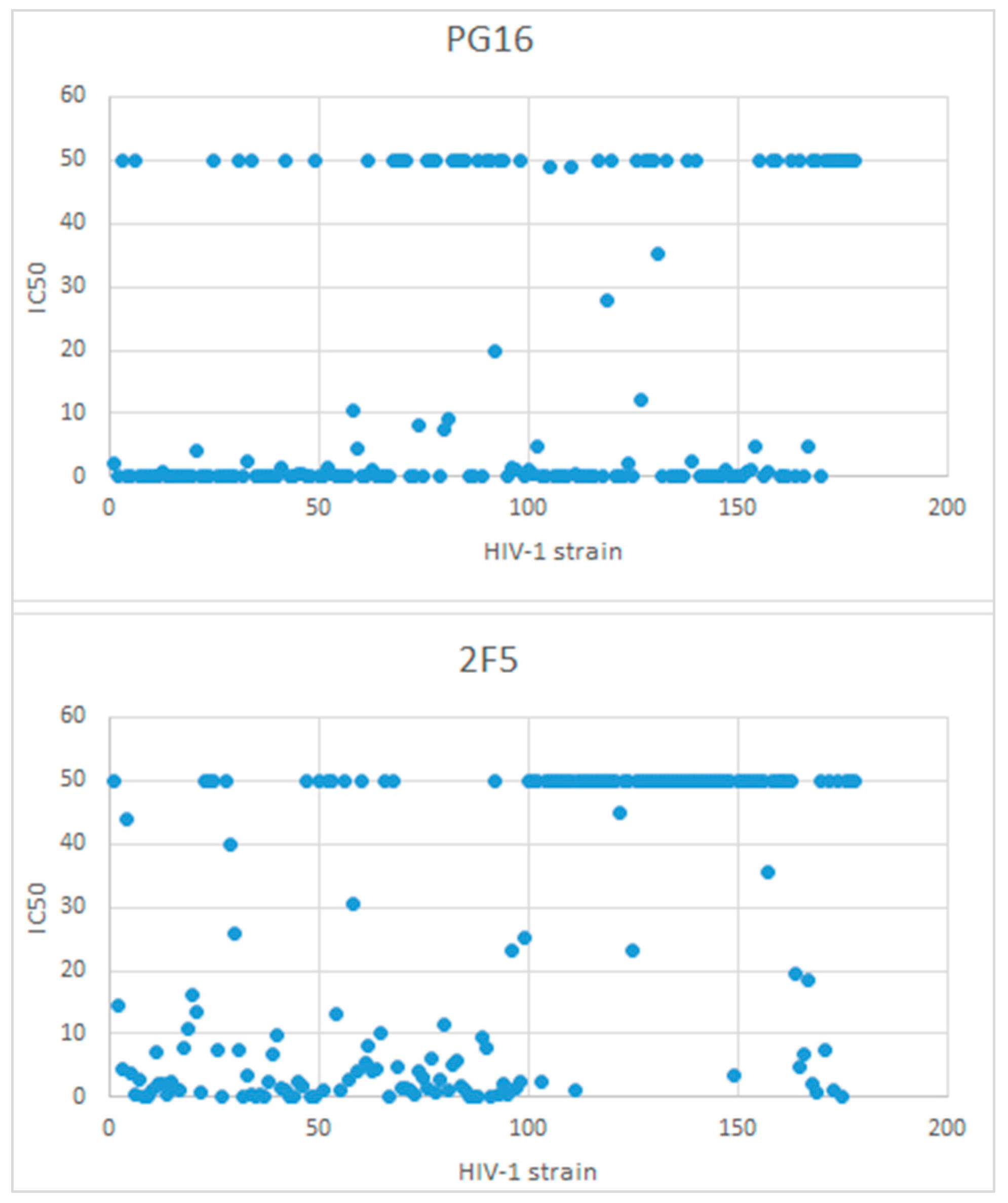{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIV-1 Strain | 2F5 | VRC01 | NIH45-46 | 3BNC117 | PG9 | PG16 |
|---|---|---|---|---|---|---|
| 0260.v5.c36 | 50 | 0.529 | 0.397 | 0.2 | 2.18 | 2.1 |
| 0330.v4.c3 | 14.6 | 0.064 | 0.049 | 0.013 | 0.018 | 0.006 |
| 0439.v5.c1 | 4.43 | 0.052 | 0.185 | 0.215 | 50 | 50 |
| 3415.v1.c1 | 43.9 | 0.092 | 0.082 | 0.094 | 0.149 | 0.036 |
| 3718.v3.c11 | 3.88 | 0.218 | 0.871 | 50 | 0.05 | 0.019 |
| 398-F1_F6_20 | 0.28 | 0.058 | 0.157 | 0.071 | 50 | 50 |
| BB201.B42 | 2.92 | 0.343 | 0.303 | 3.35 | 0.014 | 0.003 |
| BB539.2B13 | 0.136 | 0.094 | 0.022 | 0.033 | 0.106 | 0.012 |
| BI369.9A | 0.249 | 0.149 | 0.043 | 0.02 | 0.029 | 0.007 |
| BS208.B1 | 1.1 | 0.029 | 0.006 | 0.002 | 0.031 | 0.004 |
| KER2008.12 | 6.98 | 0.563 | 0.567 | 0.248 | 0.017 | 0.006 |
| KER2018.11 | 2.01 | 0.07 | 0.828 | 0.417 | 0.001 | 0.001 |
| KNH1209.18 | 2.24 | 0.087 | 0.246 | 0.04 | 0.367 | 0.678 |
| MB201.A1 | 0.436 | 0.237 | 0.165 | 0.464 | 0.024 | 0.001 |
| MB539.2B7 | 2.49 | 0.544 | 0.402 | 0.087 | 0.058 | 0.025 |
| MI369.A5 | 1.44 | 0.162 | 0.074 | 0.033 | 0.058 | 0.011 |
| MS208.A1 | 1.1 | 0.147 | 0.09 | 0.019 | 0.071 | 0.047 |
| Q168.a2 | 7.83 | 0.14 | 0.138 | 0.05 | 0.106 | 0.031 |
| Q23.17 | 10.8 | 0.086 | 0.106 | 0.017 | 0.007 | 0.002 |
| Q259.17 | 16.1 | 0.051 | 0.046 | 0.017 | 0.045 | 0.028 |
| Q461.e2 | 13.4 | 0.41 | 0.212 | 0.069 | 3.01 | 4.11 |
| Q769.d22 | 0.609 | 0.015 | 0.013 | 0.007 | 0.007 | 0.01 |
| Q769.h5 | 50 | 0.014 | 0.019 | 0.006 | 0.002 | 0.002 |
| Q842.d12 | 50 | 0.006 | 0.015 | 0.002 | 0.005 | 0.001 |
| QH209.14M.A2 | 50 | 0.024 | 0.011 | 0.008 | 50 | 50 |
| RW020.2 | 7.55 | 0.303 | 0.144 | 0.02 | 0.103 | 0.07 |
| UG037.8 | 0.202 | 0.035 | 0.056 | 0.02 | 0.021 | 0.001 |
| 3301.V1.C24 | 50 | 0.084 | 0.055 | 0.046 | 0.281 | 0.023 |
| 6540.v4.c1 | 40 | 50 | 50 | 50 | 0.035 | 0.017 |
| 6545.V4.C1 | 26 | 50 | 50 | 50 | 0.095 | 0.068 |
| 0815.V3.C3 | 7.37 | 0.036 | 0.055 | 0.018 | 50 | 50 |
| 6095.V1.C10 | 0.147 | 0.464 | 0.601 | 0.096 | 0.242 | 0.023 |
| 3468.V1.C12 | 3.51 | 0.04 | 0.104 | 0.073 | 2.09 | 2.38 |
| 620345.c1 | 0.455 | 50 | 50 | 50 | 0.393 | 50 |
| C1080.c3 | 0.056 | 1.5 | 0.539 | 0.096 | 0.004 | 0.001 |
| C2101.c1 | 0.344 | 0.097 | 2.38 | 0.064 | 0.026 | 0.009 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buiu, C.; Putz, M.V.; Avram, S. Learning the Relationship between the Primary Structure of HIV Envelope Glycoproteins and Neutralization Activity of Particular Antibodies by Using Artificial Neural Networks. Int. J. Mol. Sci. 2016, 17, 1710. https://doi.org/10.3390/ijms17101710
Buiu C, Putz MV, Avram S. Learning the Relationship between the Primary Structure of HIV Envelope Glycoproteins and Neutralization Activity of Particular Antibodies by Using Artificial Neural Networks. International Journal of Molecular Sciences. 2016; 17(10):1710. https://doi.org/10.3390/ijms17101710
Chicago/Turabian StyleBuiu, Cătălin, Mihai V. Putz, and Speranta Avram. 2016. "Learning the Relationship between the Primary Structure of HIV Envelope Glycoproteins and Neutralization Activity of Particular Antibodies by Using Artificial Neural Networks" International Journal of Molecular Sciences 17, no. 10: 1710. https://doi.org/10.3390/ijms17101710
APA StyleBuiu, C., Putz, M. V., & Avram, S. (2016). Learning the Relationship between the Primary Structure of HIV Envelope Glycoproteins and Neutralization Activity of Particular Antibodies by Using Artificial Neural Networks. International Journal of Molecular Sciences, 17(10), 1710. https://doi.org/10.3390/ijms17101710