
Cx43 Mediates Resistance against MPP+-Induced Apoptosis in SH-SY5Y Neuroblastoma Cells via Modulating the Mitochondrial Apoptosis Pathway



Abstract
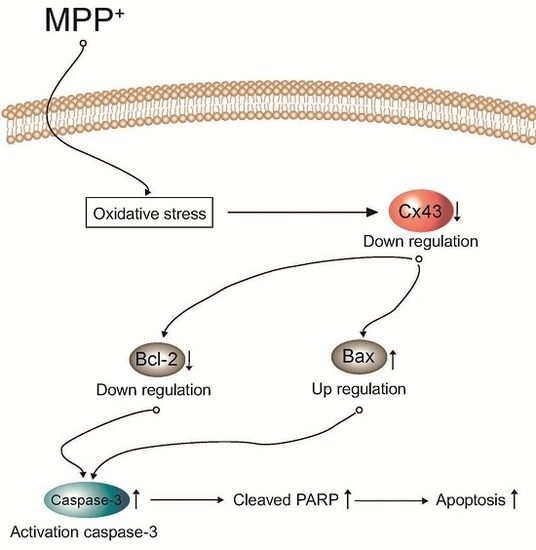
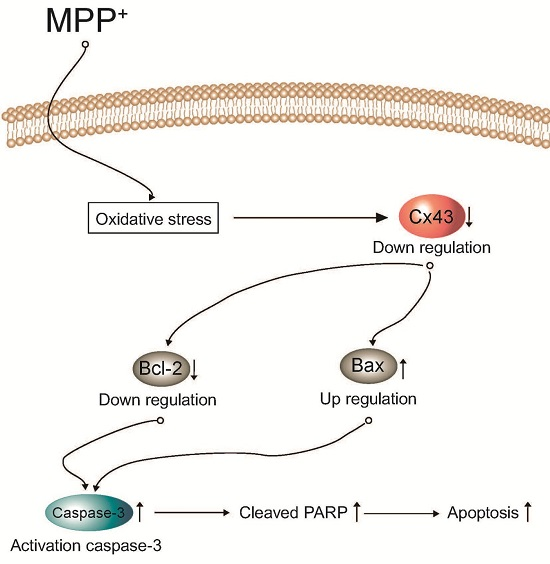
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
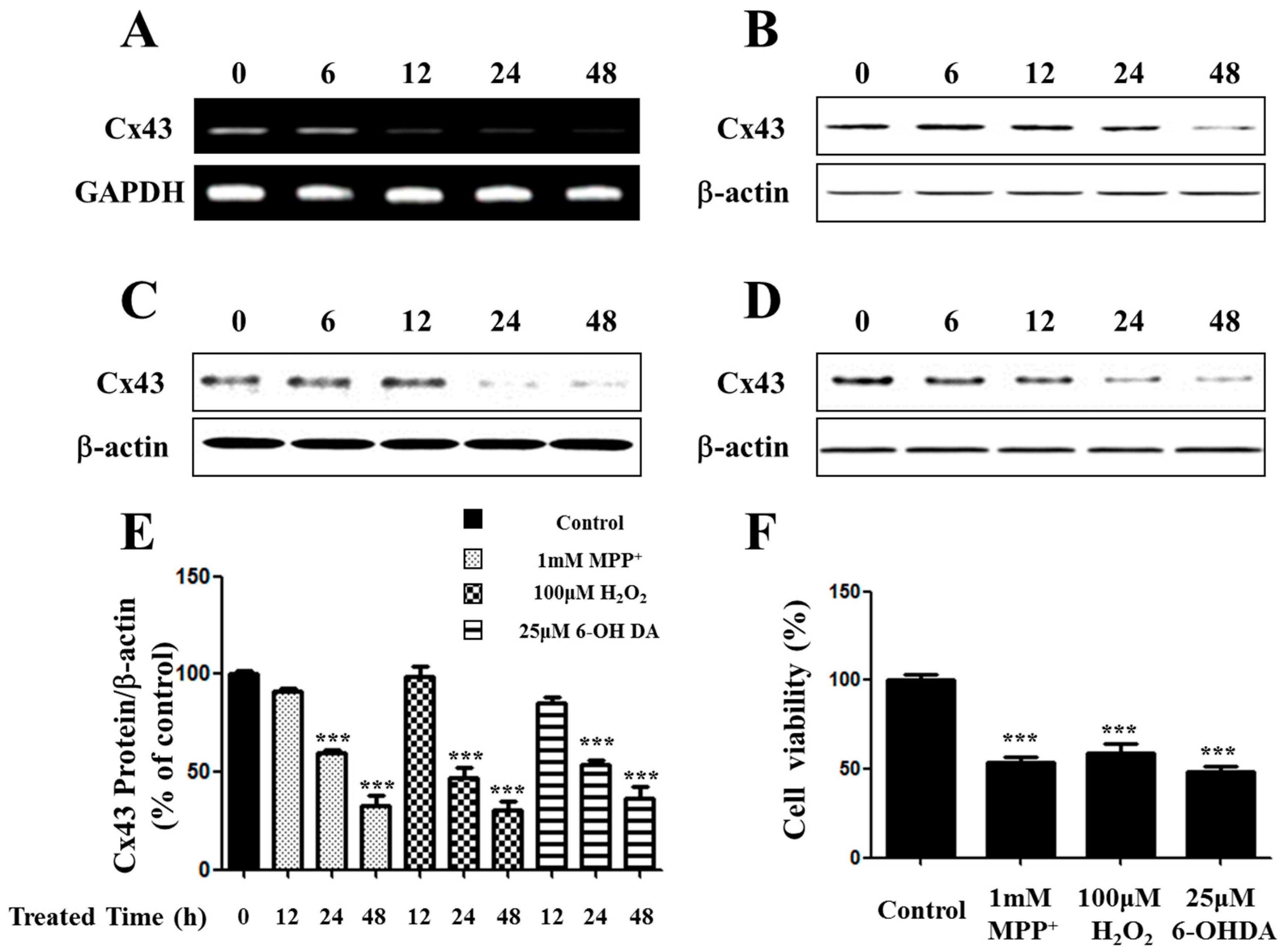
2.1. Expression of Connexin 43 (Cx43) Is Suppressed in Apoptotic SH-SY5Y Cells
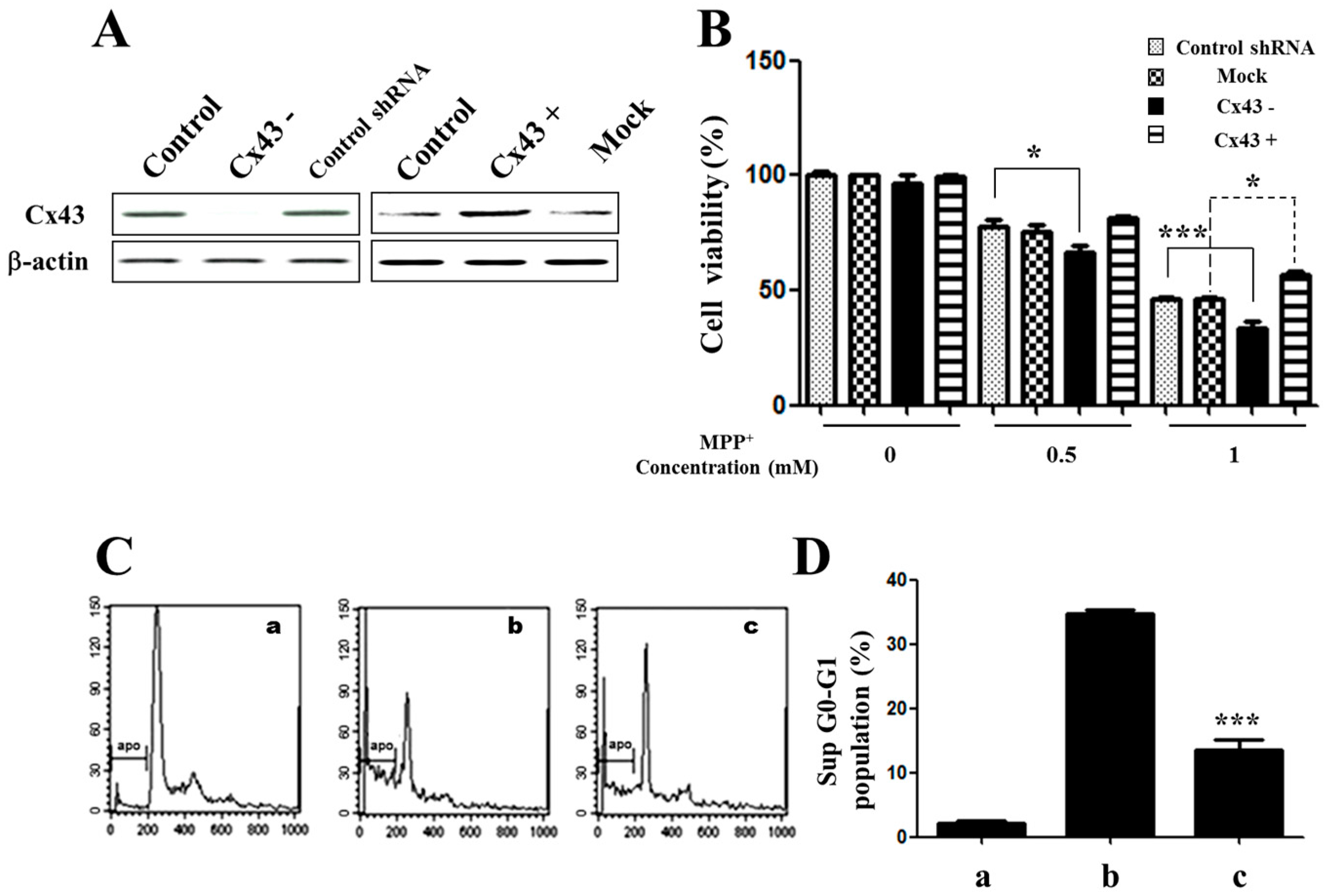
2.2. Knockdown of Cx43 Increases Sensitivity to MPP+ While Overexpression of Cx43 Reduces MPP+-Induced Apoptosis in SH-SY5Y Cells
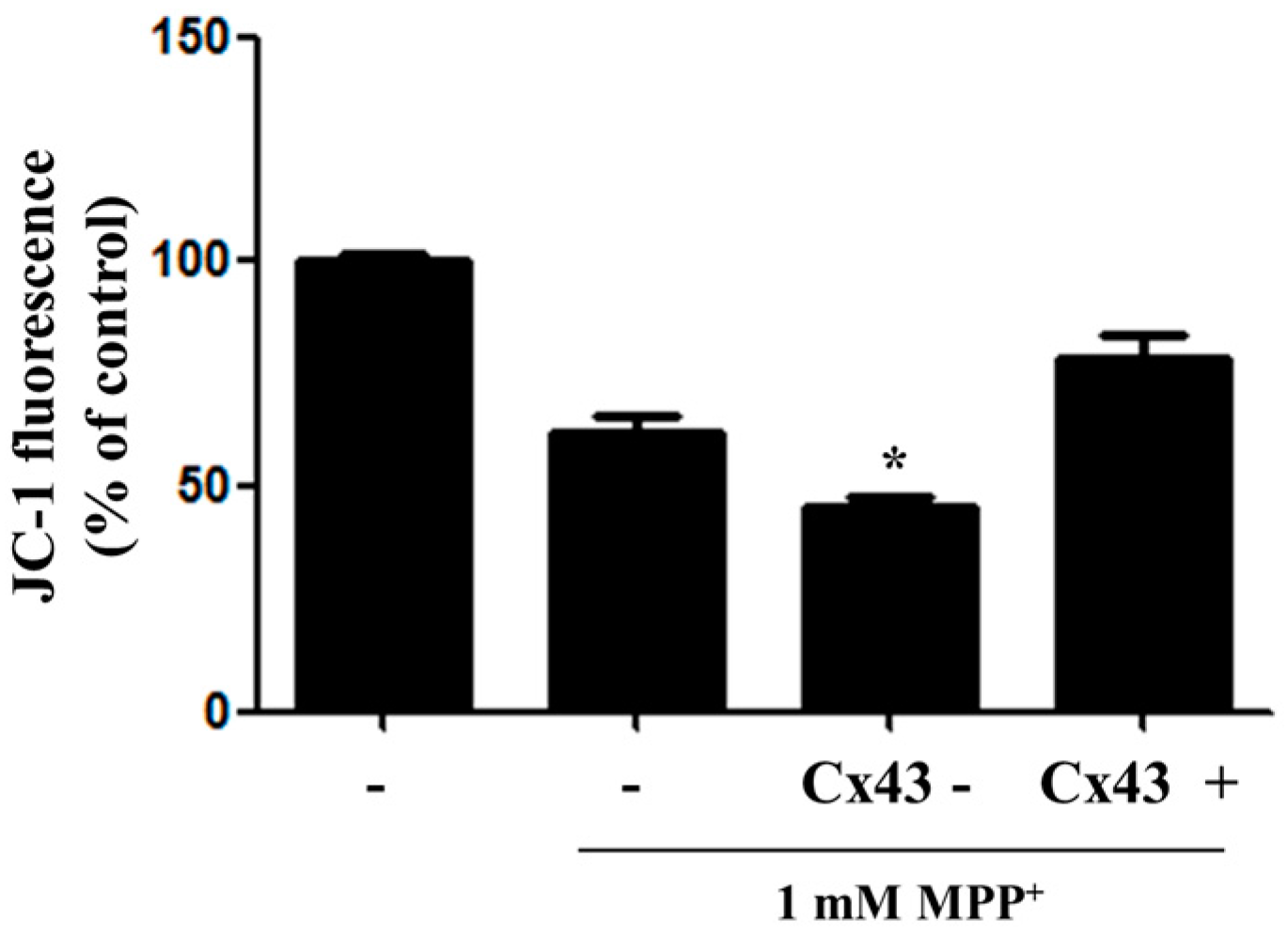
2.3. Overexpression of Cx43 Inhibits MPP+-Induced Cell Death by Increasing Mitochondrial Membrane Potential (∆Ψm) Mitochondria are Primary Target in Apoptosis
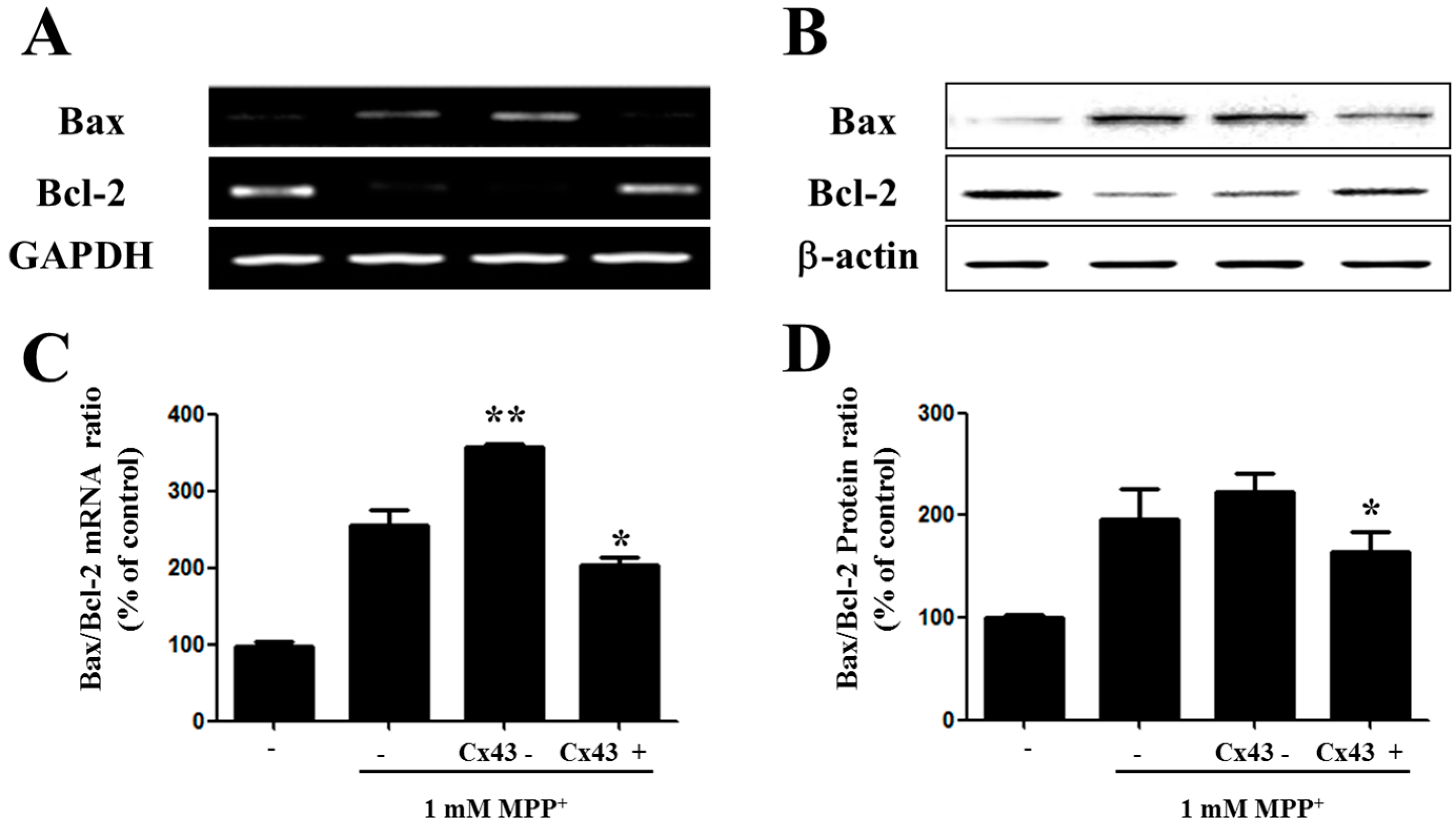
2.4. Bax/Bcl-2 Ratio Is Increased in SH-SY5YCells with Reduced Cx43 Level after MPP+ Treatment
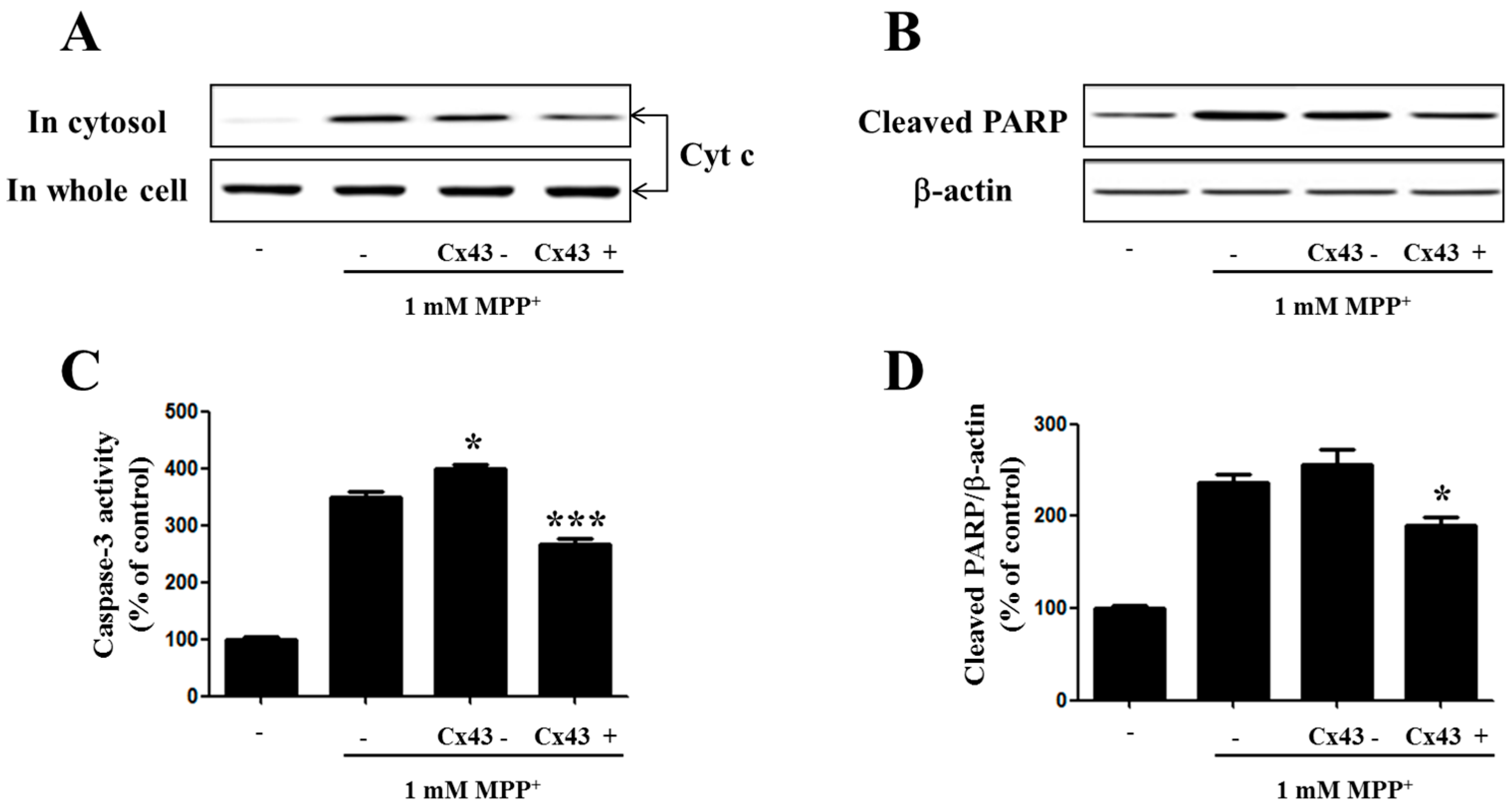
2.5. Cx43 Prevents Cytochrome C Release, Caspase-3 Activity, and PARP Proteolysis by Mitochondria
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cx43 Expression Plasmid Construction
4.3. Cell Culture and Transfection
4.4. Assessment of Cell Viability
4.5. Isolation of Total RNA and Expression Analysis
4.6. Immunoblot Analysis
4.7. Caspase-3 Activity Assay
4.8. Flow Cytometric Detection of Apoptotic Cells
4.9. Mitochondrial Membrane Potential (ΔΨm) Measurement
4.10. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Steller, H. Mechanisms and genes of cellular suicide. Science 1995, 267, 1445–1449. [Google Scholar] [CrossRef] [PubMed]
- Savill, J. Apoptosis in disease. Eur. J. Clin. Investig. 1994, 24, 715–723. [Google Scholar] [CrossRef]
- Oppenheim, R.W. Cell death during development of the nervous system. Annu. Rev. Neurosci. 1991, 14, 453–501. [Google Scholar] [CrossRef] [PubMed]
- Naruse, I.; Keino, H. Apoptosis in the developing CNS. Prog. Neurobiol. 1995, 47, 135–155. [Google Scholar] [CrossRef]
- Tanner, C.M.; Ottman, R.; Goldman, S.M.; Ellenberg, J.; Chan, P.; Mayeux, R.; Langston, J.W. Parkinson disease in twins: An etiologic study. JAMA 1999, 281, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Allam, M.F.; del Castillo, A.S.; Navajas, R.F. Parkinson’s disease risk factors: Genetic, environmental, or both? Neurol. Res. 2005, 27, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Singer, T.P.; Ramsay, R.R. Mechanism of the neurotoxicity of MPTP. An update. FEBS Lett. 1990, 274, 1–8. [Google Scholar] [PubMed]
- Eberhardt, O.; Schulz, J.B. Apoptotic mechanisms and antiapoptotic therapy in the MPTP model of Parkinson’s disease. Toxicol. Lett. 2003, 139, 135–151. [Google Scholar] [CrossRef]
- Przedborski, S.; Jackson-Lewis, V. Mechanisms of MPTP toxicity. Mov. Disord. 1998, 13, 35–38. [Google Scholar] [PubMed]
- Ogawa, T.; Hayashi, T.; Tokunou, M.; Nakachi, K.; Trosko, J.E.; Chang, C.C.; Yorioka, N. Suberoylanilide hydroxamic acid enhances gap junctional intercellular communication via acetylation of histone containing connexin 43 gene locus. Cancer Res. 2005, 65, 9771–9778. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Swanson, R.A. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia 2000, 32, 1–14. [Google Scholar] [CrossRef]
- Ransom, B.R.; Brown, A.M. Intracellular Ca2+ release and ischemic axon injury: The Trojan horse is back. Neuron 2003, 40, 2–4. [Google Scholar] [CrossRef]
- Decrock, E.; Vinken, M.; de Vuyst, E.; Krysko, D.V.; D’Herde, K.; Vanhaecke, T.; Vandenabeele, P.; Rogiers, V.; Leybaert, L. Connexin-related signaling in cell death: To live or let die? Cell Death Differ. 2009, 16, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Sin, W.C.; Crespin, S.; Mesnil, M. Opposing roles of connexin43 in glioma progression. Biochim. Biophys. Acta 2012, 1818, 2058–2067. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.I.; Li, W.; Hertzberg, E.L.; Marotta, C.A. Elevated connexin43 immunoreactivity at sites of amyloid plaques in Alzheimer’s disease. Brain Res. 1996, 717, 173–178. [Google Scholar] [CrossRef]
- Vis, J.C.; Nicholson, L.F.; Faull, R.L.; Evans, W.H.; Severs, N.J.; Green, C.R. Connexin expression in Huntington’s diseased human brain. Cell Biol. Int. 1998, 22, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Haupt, C.; Witte, O.W.; Frahm, C. Temporal profile of connexin 43 expression after photothrombotic lesion in rat brain. Neuroscience 2007, 144, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Kameritsch, P.; Khandoga, N.; Pohl, U.; Pogoda, K. Gap junctional communication promotes apoptosis in a connexin-type-dependent manner. Cell Death Dis. 2013, 4, e584. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C. Does oxidative stress participate in nerve cell death in Parkinson’s disease? Eur. Neurol. 1993, 33, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999, 341, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Vanden Hoek, T.L.; Becker, L.B.; Shao, Z.; Li, C.; Schumacker, P.T. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J. Biol. Chem. 1998, 273, 18092–18098. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M. Bax, Bid and the permeabilization of the mitochondrial outer membrane in apoptosis. Curr. Opin. Cell Biol. 2000, 12, 414–419. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Bhalla, K.; Kim, C.N.; Ibrado, A.M.; Cai, J.; Peng, T.I.; Jones, D.P.; Wang, X. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science 1997, 275, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef]
- Cryns, V.; Yuan, J. Proteases to die for. Genes Dev. 1998, 12, 1551–1570. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Hunot, S.; Michel, P.P.; Muriel, M.P.; Vyas, S.; Faucheux, B.A.; Mouatt-Prigent, A.; Turmel, H.; Srinivasan, A.; Ruberg, M.; et al. Caspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2875–2880. [Google Scholar] [CrossRef] [PubMed]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J.P.; Palmer, P.E.; Helm, G.A.; Tuttle, J.B. MPP+ induced apoptotic cell death in SH-SY5Y neuroblastoma cells: An electron microscope study. J. Neurosci. Res. 1997, 48, 226–237. [Google Scholar] [CrossRef]
- Sherer, T.B.; Betarbet, R.; Greenamyre, J.T. Environment, mitochondria, and Parkinson’s disease. Neuroscientist 2002, 8, 192–197. [Google Scholar] [PubMed]
- Choi, S.A.; Evidente, V.G.; Caviness, J.N. Comparing cerebral white matter lesion burdens between Parkinson’s disease with and without dementia. J. Mov. Disord. 2010, 3, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Dipasquale, B.; Marini, A.M.; Youle, R.J. Apoptosis and DNA degradation induced by 1-methyl-4-phenylpyridinium in neurons. Biochem. Biophys. Res. Commun. 1991, 181, 1442–1448. [Google Scholar] [CrossRef]
- Gerlach, M.; Riederer, P.; Przuntek, H.; Youdim, M.B. MPTP mechanisms of neurotoxicity and their implications for Parkinson’s disease. Eur. J. Pharmacol. 1991, 208, 273–286. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Parks, J.K.; Miller, S.W.; Tuttle, J.B.; Trimmer, P.A.; Sheehan, J.P.; Bennett, J.P., Jr.; Davis, R.E.; Parker, W.D., Jr. Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann. Neurol. 1996, 40, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Nicotra, A.; Parvez, S. Apoptotic molecules and MPTP-induced cell death. Neurotoxicol. Teratol. 2002, 24, 599–605. [Google Scholar] [CrossRef]
- Duan, L.; Yuan, H.; Su, C.J.; Liu, Y.Y.; Rao, Z.R. Ultrastructure of junction areas between neurons and astrocytes in rat supraoptic nuclei. World J. Gastroenterol. 2004, 10, 117–121. [Google Scholar] [PubMed]
- Pereda, A.E. Electrical synapses and their functional interactions with chemical synapses. Nat. Rev. Neurosci. 2014, 15, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Giaume, C.; Theis, M. Pharmacological and genetic approaches to study connexin-mediated channels in glial cells of the central nervous system. Brain Res. Rev. 2010, 63, 160–176. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.J.; Simek, J.; Laird, D.W. Mechanisms linking connexin mutations to human diseases. Cell Tissue Res. 2015, 360, 701–721. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Weigel, H.; Cotrina, M.L.; Liu, S.; Bueno, E.; Hansen, A.J.; Hansen, T.W.; Goldman, S.; Nedergaard, M. Gap-junction-mediated propagation and amplification of cell injury. Nat. Neurosci. 1998, 1, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dorado, D.; Rodriguez-Sinovas, A.; Ruiz-Meana, M. Gap junction-mediated spread of cell injury and death during myocardial ischemia-reperfusion. Cardiovasc. Res. 2004, 61, 386–401. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Liu, Y.G.; Lin, Y.; Fan, Y.; Boynton, A.; Yang, D.; Huang, R.P. Enhanced apoptosis under low serum conditions in human glioblastoma cells by connexin 43 (Cx43). Mol. Carcinog. 2001, 32, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Nakase, T.; Sohl, G.; Theis, M.; Willecke, K.; Naus, C.C. Increased apoptosis and inflammation after focal brain ischemia in mice lacking connexin43 in astrocytes. Am. J. Pathol. 2004, 164, 2067–2075. [Google Scholar] [CrossRef]
- Goldberg, G.S.; Lampe, P.D.; Nicholson, B.J. Selective transfer of endogenous metabolites through gap junctions composed of different connexins. Nat. Cell Biol. 1999, 1, 457–459. [Google Scholar] [CrossRef] [PubMed]
- Cottin, S.; Ghani, K.; de Campos-Lima, P.O.; Caruso, M. Gemcitabine intercellular diffusion mediated by gap junctions: New implications for cancer therapy. Mol. Cancer 2010, 9, 141. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Haider, H.; Porollo, A.; Ashraf, M. Mitochondria-specific transgenic overexpression of connexin-43 simulates preconditioning-induced cytoprotection of stem cells. Cardiovasc. Res. 2010, 88, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Meana, M.; Rodriguez-Sinovas, A.; Cabestrero, A.; Boengler, K.; Heusch, G.; Garcia-Dorado, D. Mitochondrial connexin43 as a new player in the pathophysiology of myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2008, 77, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Datki, Z.; Juhász, A.; Gálfi, M.; Soós, K.; Papp, R.; Zádori, D.; Penke, B. Method for measuring neurotoxicity of aggregating polypeptides with the MTT assay on differentiated neuroblastoma cells. Brain Res. Bull. 2003, 62, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kim, I.S.; More, S.V.; Kim, B.W.; Bahk, Y.Y.; Choi, D.K. Gastrodin protects apoptotic dopaminergic neurons in a toxin-induced Parkinson’s disease model. Evid. Based Complement. Altern. Med. 2013, 2013, 514095. [Google Scholar] [CrossRef] [PubMed]
- Telford, W.G.; King, L.E.; Fraker, P.J. Evaluation of glucocorticoid-induced DNA fragmentation in mouse thymocytes by flow cytometry. Cell Prolif. 1991, 24, 447–459. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, I.-S.; Ganesan, P.; Choi, D.-K. Cx43 Mediates Resistance against MPP+-Induced Apoptosis in SH-SY5Y Neuroblastoma Cells via Modulating the Mitochondrial Apoptosis Pathway. Int. J. Mol. Sci. 2016, 17, 1819. https://doi.org/10.3390/ijms17111819
Kim I-S, Ganesan P, Choi D-K. Cx43 Mediates Resistance against MPP+-Induced Apoptosis in SH-SY5Y Neuroblastoma Cells via Modulating the Mitochondrial Apoptosis Pathway. International Journal of Molecular Sciences. 2016; 17(11):1819. https://doi.org/10.3390/ijms17111819
Chicago/Turabian StyleKim, In-Su, Palanivel Ganesan, and Dong-Kug Choi. 2016. "Cx43 Mediates Resistance against MPP+-Induced Apoptosis in SH-SY5Y Neuroblastoma Cells via Modulating the Mitochondrial Apoptosis Pathway" International Journal of Molecular Sciences 17, no. 11: 1819. https://doi.org/10.3390/ijms17111819
APA StyleKim, I. -S., Ganesan, P., & Choi, D. -K. (2016). Cx43 Mediates Resistance against MPP+-Induced Apoptosis in SH-SY5Y Neuroblastoma Cells via Modulating the Mitochondrial Apoptosis Pathway. International Journal of Molecular Sciences, 17(11), 1819. https://doi.org/10.3390/ijms17111819