
Effect of High-Carbohydrate Diet on Plasma Metabolome in Mice with Mitochondrial Respiratory Chain Complex III Deficiency

 , and
, and
Abstract
:
1. Introduction
2. Results
2.1. Reduced Survival and No Effect on Blood Glucose or Weight by High Carbohydrate Diet (HCD) Feeding
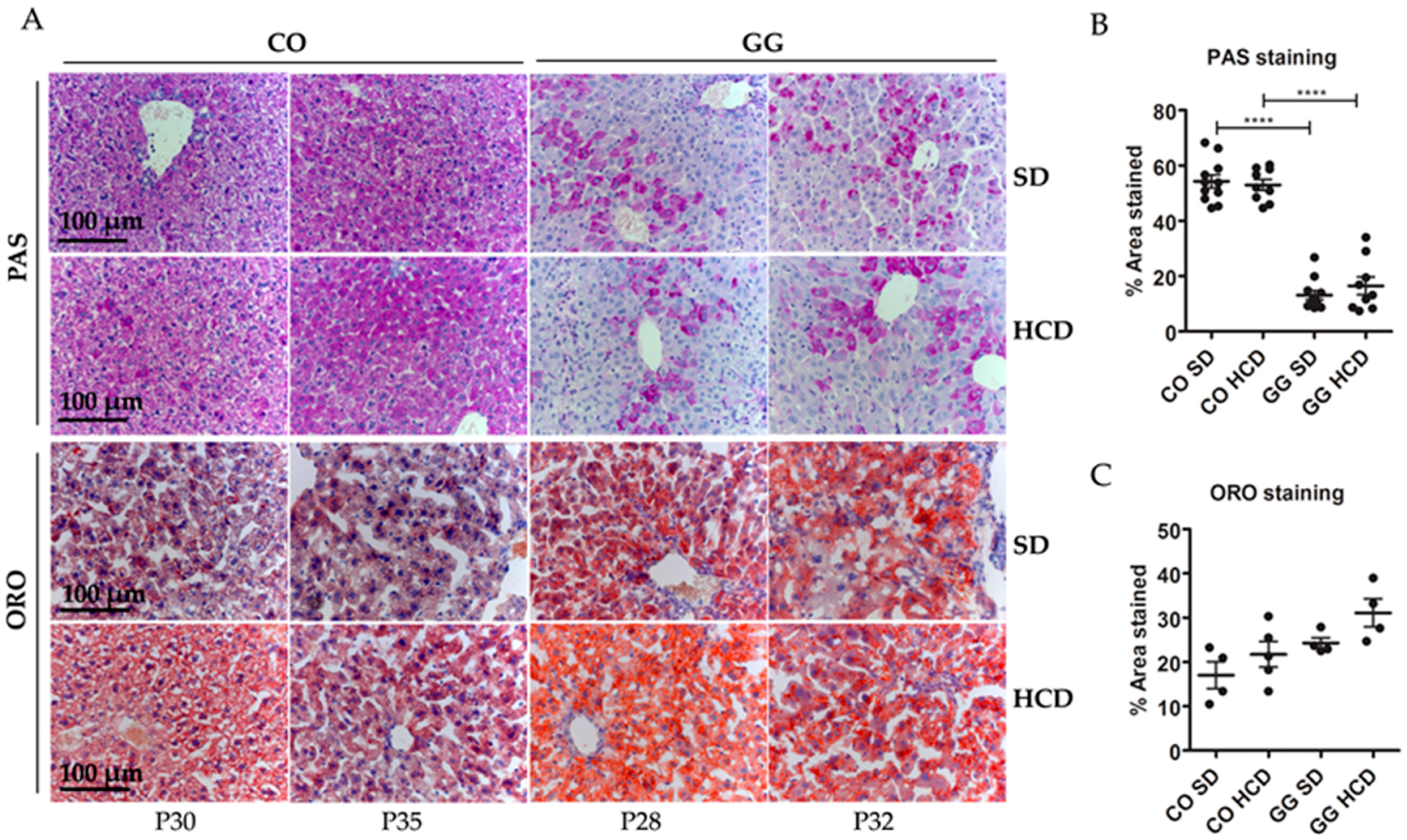
2.2. No Effect on Hepatic Glycogen Depletion or Fat Accumulation by HCD Feeding
2.3. No Effect on Respiratory Chain (RC) Complex Assembly or Respiration in Liver Mitochondria by HCD Feeding
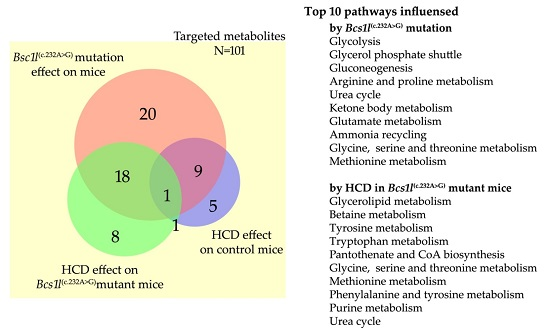
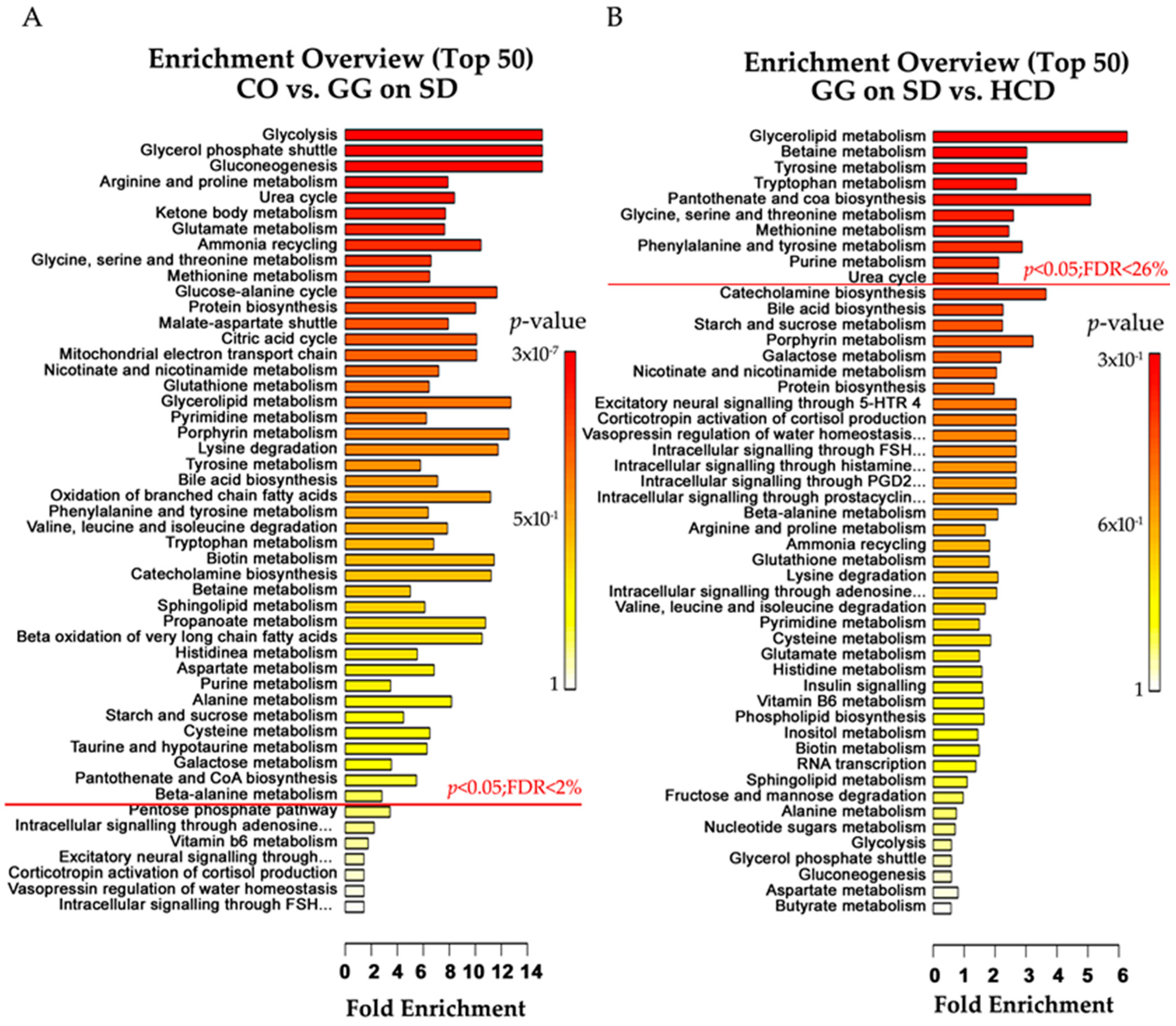
2.4. Altered Plasma Metabolome in Bcs1l Mutant Mice and Partial Normalization of Amino Acid Metabolism and Urea Cycle by HCD Feeding
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Ethics Statement
4.3. Tissue Sampling
4.4. Histology
4.5. Respiratory Chain Assessment in Liver Mitochondria by Respirometry and BNGE
4.6. Metabolomics Analysis from Plasma
4.7. Statistics
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Honzik, T.; Tesarova, M.; Magner, M.; Mayr, J.; Jesina, P.; Vesela, K.; Wenchich, L.; Szentivanyi, K.; Hansikova, H.; Sperl, W.; et al. Neonatal onset of Mitochondrial disorders in 129 patients: Clinical and laboratory characteristics and a new approach to diagnosis. J. Inherit. Metab. Dis. 2012, 35, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Sokol, R.J. Liver disease in mitochondrial disorders. Semin. Liver Dis. 2007, 27, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Burd, R.S.; Whalen, T.V. Evaluation of the child with suspected appendicitis. Pediatr. Ann. 2001, 30, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Arroyo, M.A.; Hualde, J.; Ayechu, A.; de Meirleir, L.; Seneca, S.; Nadal, N.; Briones, P. Clinical and biochemical spectrum of mitochondrial complex III deficiency caused by mutations in the BCS1L gene. Clin. Genet. 2009, 75, 585–587. [Google Scholar] [PubMed]
- Visapää, I.; Fellman, V.; Vesa, J.; Dasvarma, A.; Hutton, J.L.; Kumar, V.; Payne, G.S.; Makarow, M.; van Coster, R.; Taylor, R.W.; et al. GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L. Am. J. Hum. Genet. 2002, 71, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Fellman, V.; Rapola, J.; Pihko, H.; Varilo, T.; Raivio, K.O. Iron-overload disease in infants involving fetal growth retardation, lactic acidosis, liver haemosiderosis, and aminoaciduria. Lancet 1998, 351, 490–493. [Google Scholar] [CrossRef]
- Fellman, V.; Kotarsky, H. Mitochondrial hepatopathies in the newborn period. Semin. Fetal Neonatal Med. 2011, 16, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Levéen, P.; Kotarsky, H.; Mörgelin, M.; Karikoski, R.; Elmér, E.; Fellman, V. The GRACILE mutation introduced into Bcs1l causes postnatal complex III deficiency: A viable mouse model for mitochondrial hepatopathy. Hepatology 2011, 53, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Paoli, A.; Bianco, A.; Damiani, E.; Bosco, G. Review Article Ketogenic Diet in Neuromuscular and Neurodegenerative Diseases. BioMed Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nyman, L.R.; Tian, L.; Hamm, D.A.; Schoeb, T.R.; Gower, B.A.; Nagy, T.R.; Wood, P. A Long term effects of high fat or high carbohydrate diets on glucose tolerance in mice with heterozygous carnitine palmitoyltransferase-1a (CPT-1a) deficiency: Diet influences on CPT1a deficient mice. Nutr. Diabetes 2011, 1, e14. [Google Scholar] [CrossRef] [PubMed]
- Douris, N.; Melman, T.; Pecherer, J.M.; Pissios, P.; Flier, J.S.; Cantley, L.C.; Locasale, J.W.; Maratos-Flier, E. Adaptive changes in amino acid metabolism permit normal longevity in mice consuming a low-carbohydrate ketogenic diet. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2056–2065. [Google Scholar] [CrossRef] [PubMed]
- Bénit, P.; Rustin, P. Changing the diet to make more mitochondria and protect the heart. Circ. Res. 2012, 110, 1047–1048. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Z.; Chen, J.K.; Li, Z.P.; Zhao, T.; Ni, M.; Li, D.J.; Jiang, C.L.; Shen, F.M. High-salt diet enhances hippocampal oxidative stress and cognitive impairment in mice. Neurobiol. Learn. Mem. 2014, 114, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Sumiyoshi, M.; Sakanaka, M.; Kimura, Y. Chronic intake of high-fat and high-sucrose diets differentially affects glucose intolerance in mice. J. Nutr. 2006, 136, 582–587. [Google Scholar] [PubMed]
- Davoudi, M.; Kotarsky, H.; Hansson, E.; Fellman, V. Complex I function and supercomplex formation are preserved in liver mitochondria despite progressive complex III deficiency. PLoS ONE 2014, 9, e86767. [Google Scholar] [CrossRef] [PubMed]
- Jessy, J.; Mans, A.M.; DeJoseph, M.R.; Hawkins, R.A. Hyperammonaemia causes many of the changes found after portacaval shunting. Biochem. J. 1990, 272, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Kotarsky, H.; Keller, M.; Davoudi, M.; Levéen, P.; Karikoski, R.; Enot, D.P.; Fellman, V. Metabolite profiles reveal energy failure and impaired β-oxidation in liver of mice with complex III deficiency due to a BCS1L mutation. PLoS ONE 2012, 7, e41156. [Google Scholar] [CrossRef] [PubMed]
- Fryburg, D.A.; Barrett, E.J.; Louard, R.J.; Gelfand, R.A. Effect of starvation on human muscle protein metabolism and its response to insulin. Am. J. Physiol. Metab. 1990, 259, E477–E482. [Google Scholar]
- Dejong, C.; van de Poll, M.; Soeters, P.; Jalan, R.; Damink, S. Aromatic amino acid metabolism during liver failure. J. Nutr. 2007, 137, 1579S–1585S. [Google Scholar] [PubMed]
- Holecek, M. Ammonia and amino acid profiles in liver cirrhosis: Effects of variables leading to hepatic encephalopathy. Nutrition 2015, 31, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.H.; Parikh, S.; Falk, M.J.; Saneto, R.P.; Wolf, N.I.; Darin, N.; Wong, L.J.; Cohen, B.H.; Naviaux, R.K. The in-depth evaluation of suspected mitochondrial disease. Mol. Genet. Metab. 2008, 94, 16–37. [Google Scholar] [CrossRef] [PubMed]
- Felig, P. The glucose-alanine cycle. Metabolism 1973, 22, 179–207. [Google Scholar] [CrossRef]
- Clarke, C.; Xiao, R.; Place, E.; Zhang, Z.; Sondheimer, N.; Bennett, M.; Yudkoff, M.; Falk, M.J. Mitochondrial respiratory chain disease discrimination by retrospective cohort analysis of blood metabolites. Mol. Genet. Metab. 2013, 110, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Victor, W.; Rodwell, P. Chapter 29. Catabolism of the Carbon Skeletons of Amino Acids. In Harper’s Illustrated Biochemistry, 29e; Murray, R.K., Bender, D.A., Botham, K.M., Kennelly, P.J., Rodwell, V.W., Weil, P.A., Eds.; The McGraw-Hill Companies: New York, NY, USA, 2012. [Google Scholar]
- Oxenkrug, G.F. Tryptophan kynurenine metabolism as a common mediator of genetic and environmental impacts in major depressive disorder: The serotonin hypothesis revisited 40 years later. Isr. J. Psychiatry Relat. Sci. 2010, 47, 56–63. [Google Scholar] [PubMed]
- Oxenkrug, G.F. Associated psychiatric and medical disorders. J. Neural Transm. 2011, 118, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Oxenkrug, G. Insulin resistance and dysregulation of tryptophan—Kynurenine and kynurenine—Nicotinamide adenine dinucleotide metabolic pathways. Mol. Neurobiol. 2013, 48, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Ocampo, J.; Ramírez-Ortega, D.; Vázquez Cervantes, G.I.; Pineda, B.; Montes de Oca Balderas, P.; González-Esquivel, D.; Sánchez-Chapul, L.; Lugo-Huitrón, R.; Silva-Adaya, D.; Ríos, C.; et al. Mitochondrial dysfunction related to cell damage induced by 3-hydroxykynurenine and 3-hydroxyanthranilic acid: Non-dependent-effect of early reactive oxygen species production. Neurotoxicology 2015, 50, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Psihogios, N.G.; Kalaitzidis, R.G.; Dimou, S.; Seferiadis, K.I.; Siamopoulos, K.C.; Bairaktari, E.T. Evaluation of tubulointerstitial lesions’ severity in patients with glomerulonephritides: An NMR-based metabonomic study. J. Proteome Res. 2007, 6, 3760–3770. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xiong, Z.; Li, J.; Zheng, S.; Huo, T.; Li, F. Metabonomic study on “Kidney-Yang Deficiency syndrome” and intervention effects of Rhizoma Drynariae extracts in rats using ultra performance liquid chromatography coupled with mass spectrometry. Talanta 2011, 83, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, A.; Nakashima, E.; Sugimoto, M.; Akiyama, S.; Sato, W.; Maruyama, S.; Matsuo, S.; Tomita, M.; Yuzawa, Y.; Soga, T. Metabolic profiling reveals new serum biomarkers for differentiating diabetic nephropathy. Anal. Bioanal. Chem. 2012, 404, 3101–3109. [Google Scholar] [CrossRef] [PubMed]
- Fleck, C.; Schweitzer, F.; Karge, E.; Busch, M.; Stein, G. Serum concentrations of asymmetric (ADMA) and symmetric (SDMA) dimethylarginine in patients with chronic kidney diseases. Clin. Chim. Acta 2003, 336, 1–12. [Google Scholar] [CrossRef]
- Schefold, J.C.; Zeden, J.P.; Fotopoulou, C.; von Haehling, S.; Pschowski, R.; Hasper, D.; Volk, H.D.; Schuett, C.; Reinke, P. Increased indoleamine 2,3-dioxygenase (IDO) activity and elevated serum levels of tryptophan catabolites in patients with chronic kidney disease: A possible link between chronic inflammation and uraemic symptoms. Nephrol. Dial. Transplant. 2009, 24, 1901–1908. [Google Scholar] [CrossRef] [PubMed]
- Abdo, A.S.; Basu, A.; Geraci, S. A managing chronic heart failure patient in chronic kidney disease. Am. J. Med. 2011, 124, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Ezgu, F.; Senaca, S.; Gunduz, M.; Tumer, L.; Hasanoglu, A.; Tiras, U.; Unsal, R.; Bakkaloglu, S.A. Severe renal tubulopathy in a newborn due to BCS1L gene mutation: Effects of different treatment modalities on the clinical course. Gene 2013, 528, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Demine, S.; Reddy, N.; Renard, P.; Raes, M.; Arnould, T. Unraveling biochemical pathways affected by mitochondrial dysfunctions using metabolomic approaches. Metabolites 2014, 4, 831–878. [Google Scholar] [CrossRef] [PubMed]
- Shiba, N.; Shimokawa, H. Chronic kidney disease and heart failure-Bidirectional close link and common therapeutic goal. J. Cardiol. 2011, 57, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Hewetson, M.; Cohen, N.D.; Love, S.; Buddington, R.K.; Holmes, W.; Innocent, G.T.; Roussel, A.J. Sucrose concentration in blood: A new method for assessment of gastric permeability in horses with gastric ulceration. J. Vet. Intern. Med. 2006, 20, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Le, K.-A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Shi, Y.H.; Le, G.W. Oxidative stress induced by high-glucose diet in liver of C57BL/6J mice and its underlying mechanism. Mol. Biol. Rep. 2010, 37, 3833–3839. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Huitrón, R.; Blanco-Ayala, T.; Ugalde-Muñiz, P.; Carrillo-Mora, P.; Pedraza-Chaverrí, J.; Silva-Adaya, D.; Maldonado, P.D.; Torres, I.; Pinzón, E.; Ortiz-Islas, E.; et al. On the antioxidant properties of kynurenic acid: Free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol. Teratol. 2011, 33, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Swick, W. Metabolism of Vitamin B6 in Rat Liver Mitochondria. J. Biol. Chem. 1957, 256, 751–764. [Google Scholar]
- Ulvik, A.; Midttun, O.; Pedersen, E.R.; Eussen, S.J.P.M.; Nygard, O.; Ueland, P.M. Evidence for increased catabolism of vitamin B-6 during systemic inflammation. Am. J. Clin. Nutr. 2014, 100, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open source platform for biological image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Roman-Garcia, P.; Quiros-Gonzalez, I.; Mottram, L.; Lieben, L.; Sharan, K.; Wangwiwatsin, A.; Tubio, J.; Lewis, K.; Wilkinson, D.; Santhanam, B.; et al. Vitamin B 12—Dependent taurine synthesis regulates growth and bone mass. J. Clin. Investig. 2014, 124, 2988–3002. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolites (mmol/L) | Control SD (n = 38) | Control HCD (n = 26) | GG SD (n = 30) | GG HCD (n = 21) |
|---|---|---|---|---|
| Glucose | 7.0 ± 0.7 | 6.9 ± 1.2 | 1.7 ± 0.6 1 | 2.4 ± 1.6 2 |
| Lactate | 4.0 ± 2.2 | 4.6 ± 1.9 | 3.5 ± 1.8 | 4.1 ± 1.2 |
| Lactate/Glucose | 0.45 ± 0.2 | 0.6 ± 0.2 | 2.0 ± 1.0 1 | 2.9 ± 2.4 2 |
| Ketones | 0.6 ± 0.3 | 0.8 ± 0.3 | 0.5 ± 0.2 | 0.7 ± 0.3 |
| Metabolites (mmol/L) | Control SD (n = 12) | Control HCD (n = 6) | GG SD (n = 6) | GG HCD (n = 6) |
|---|---|---|---|---|
| Amino acid derivatives | ||||
| Asymmetricdimethylarginine | 4.05 ± 0.7 | 3.39 ± 0.64 | 5.55 ± 1.51 1 | 4.39 ± 0.7 |
| Asparagine | 136 ± 29.8 | 144 ± 75.1 | 432 ± 164.6 1 | 311 ± 113 2 |
| Creatinine | 5.23 ± 1.2 | 5.19 ± 0.95 | 5.6 ± 1.93 | 13.2 ± 5.4 2,3 |
| Dimethylglycine | 8.03 ± 3.7 | 3.92 ± 1.9 | 30.1 ± 11.3 1 | 13.3 ± 4.8 3 |
| Glutamine | 918 ± 185 | 831 ± 65.2 | 2547 ± 754 1 | 1457 ± 363 2,3 |
| Glycine | 354 ± 116 | 246 ± 62.8 | 897 ± 229 1 | 619 ± 242 2,3 |
| Kynurenic Acid | 0.06 ± 0.03 | 0.102 ± 0.04 | 0.12 ± 0.08 | 0.57 ± 0.36 2,3 |
| Leucine | 149 ± 50.9 | 211 ± 117 | 413 ± 119 1 | 269 ± 69.9 3 |
| Phenylalanine | 43.9 ± 8.8 | 56.6 ± 13.5 | 98.0 ± 21.4 1 | 64.8 ± 16.2 3 |
| Symmetricdimethylarginine | 1.3 ± 0.26 | 1.1 ± 0.42 | 1.72 ± 0.43 | 2.3 ± 0.4 2,3 |
| Tyrosine | 55.2 ± 11.8 | 86.5 ± 23.6 | 164 ± 67.9 1 | 93.7 ± 37.9 3 |
| Neurotransmitter intermediates | ||||
| 5-Hydroxyindole-3-acetic acid | 0.23 ± 0.13 | 0.32 ± 0.14 | 0.69 ± 0.47 | 3.18 ± 1.9 2,3 |
| l-5-Hydroxytryptophan | 0.024 ± 0.008 | 0.04 ± 0.02 | 0.09 ± 0.04 1 | 0.06 ± 0.02 3 |
| Normetanephrine | 0.018 ± 0.006 | 0.02 ± 0.006 | 0.005 ± 0.002 1 | 0.02 ± 0.012 3 |
| Others | ||||
| 4-pyridoxic acid | 0.045 ± 0.03 | 0.036 ± 0.03 | 0.11 ± 0.1 | 0.32 ± 0.23 2,3 |
| Adenine | 0.006 ± 0.003 | 0.005 ± 0.001 | 0.005 ± 0.001 | 0.009 ± 0.005 |
| Adenosine | 0.05 ± 0.03 | 0.05 ± 0.02 1 | 0.08 ± 0.025 | 0.10 ± 0.05 1,2 |
| cAMP | 0.007 ± 0.003 | 0.007 ± 0.002 | 0.01 ± 0.003 | 0.015 ± 0.007 1,2 |
| Chanodeoxycholic acid | 41.7 ± 20.4 | 472 ± 717 | 1295 ± 1578 1 | 257 ± 356 |
| Cholic Acid | 9.8 ± 11.5 | 25.1 ± 38.7 | 428 ± 228.6 1 | 23.4 ± 14.24 3 |
| Cotinine | 0.011 ± 0.003 | 0.01 ± 0.005 | 0.005 ± 0.002 1 | 0.01 ± 0.005 3 |
| Glyceraldehyde | 48.9 ± 14.6 | 67 ± 21.5 | 115 ± 27.01 1 | 71.9 ± 12 3 |
| Glycocholic acid | 0.46 ± 0.14 | 0.21 ± 0.07 | 0.67 ± 0.49 2 | 0.36 ± 0.16 |
| Isobutyryl carnitine | 0.037 ± 0.01 | 0.022 ± 0.01 | 0.06 ± 0.02 1 | 0.024 ± 0.01 3 |
| Nicotinic acid | 0.11 ± 0.06 | 0.08 ± 0.04 | 0.29 ± 0.29 | 0.12 ± 0.57 |
| Ornithine | 71.8 ± 11.3 | 86.5 ± 20.7 | 297 ± 75.4 1 | 190 ± 43.8 3 |
| Pantothenic acid | 3.9 ± 1.14 | 5.4 ± 1.6 | 2.56 ± 0.73 | 5.64 ± 2.4 3 |
| Sucrose | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.5 ± 0.1 1 | 0.4 ± 0.1 2 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajendran, J.; Tomašić, N.; Kotarsky, H.; Hansson, E.; Velagapudi, V.; Kallijärvi, J.; Fellman, V. Effect of High-Carbohydrate Diet on Plasma Metabolome in Mice with Mitochondrial Respiratory Chain Complex III Deficiency. Int. J. Mol. Sci. 2016, 17, 1824. https://doi.org/10.3390/ijms17111824
Rajendran J, Tomašić N, Kotarsky H, Hansson E, Velagapudi V, Kallijärvi J, Fellman V. Effect of High-Carbohydrate Diet on Plasma Metabolome in Mice with Mitochondrial Respiratory Chain Complex III Deficiency. International Journal of Molecular Sciences. 2016; 17(11):1824. https://doi.org/10.3390/ijms17111824
Chicago/Turabian StyleRajendran, Jayasimman, Nikica Tomašić, Heike Kotarsky, Eva Hansson, Vidya Velagapudi, Jukka Kallijärvi, and Vineta Fellman. 2016. "Effect of High-Carbohydrate Diet on Plasma Metabolome in Mice with Mitochondrial Respiratory Chain Complex III Deficiency" International Journal of Molecular Sciences 17, no. 11: 1824. https://doi.org/10.3390/ijms17111824
APA StyleRajendran, J., Tomašić, N., Kotarsky, H., Hansson, E., Velagapudi, V., Kallijärvi, J., & Fellman, V. (2016). Effect of High-Carbohydrate Diet on Plasma Metabolome in Mice with Mitochondrial Respiratory Chain Complex III Deficiency. International Journal of Molecular Sciences, 17(11), 1824. https://doi.org/10.3390/ijms17111824