
The Multifaceted Roles of PI3Kγ in Hypertension, Vascular Biology, and Inflammation

Abstract
:
1. Introduction
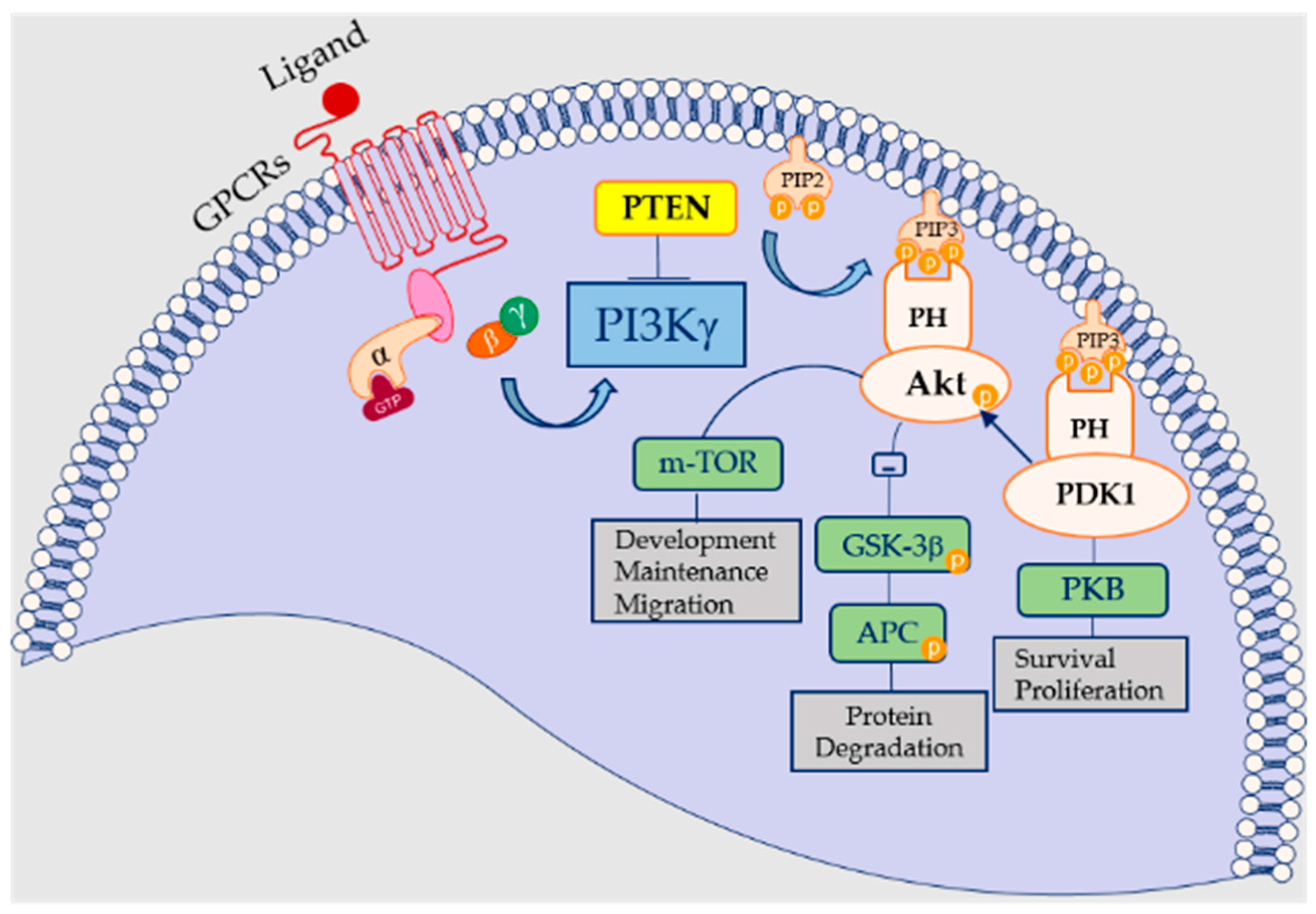
2. PI3Kγ: A Focus on Their Signaling Pathway in Cardiovascular Disease and Hypertension
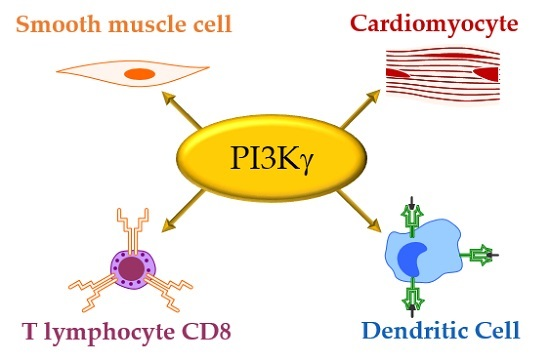
3. PI3Kγ in Immunity
4. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.; Kiger, A.A. Classes of phosphoinositide 3-kinases at a glance. J. Cell Sci. 2014, 127, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Morello, F.; Perino, A.; Hirsch, E. Phosphoinositide 3-kinase signalling in the vascular system. Cardiovasc. Res. 2009, 82, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Martin-Conte, E.L.; Hirsch, E. Phosphoinositide 3-kinase p110γ in immunity. IUBMB Life 2011, 63, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Wymann, M.P.; Björklöf, K.; Calvez, R.; Finan, P.; Thomast, M.; Trifilieff, A.; Barbier, M.; Altruda, F.; Hirsch, E.; Laffargue, M. Phosphoinositide 3-kinase gamma: A key modulator in inflammation and allergy. Biochem. Soc. Trans. 2003, 31, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Laffargue, M.; Calvez, R.; Finan, P.; Trifilieff, A.; Barbier, M.; Altruda, F.; Hirsch, E.; Wymann, M.P. Phosphoinositide 3-kinase gamma is an essential amplifier of mast cell function. Immunity 2002, 16, 441–451. [Google Scholar] [CrossRef]
- Belmonte, S.L.; Blaxall, B.C. G protein coupled receptor kinases as therapeutic targets in cardiovascular disease. Circ. Res. 2011, 109, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Kok, K.; Geering, B.; Vanhaesebroeck, B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem. Sci. 2009, 34, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Lupieri, A.; Smirnova, N.; Malet, N.; Gayral, S.; Laffargue, M. PI3K signaling in arterial diseases: Non redundant functions of the PI3K isoforms. Adv. Biol. Regul. 2015, 59, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Rommel, C. Taking PI3Kdelta and PI3Kγ one step ahead: dual active PI3KΔ/γ inhibitors for the treatment of immune-mediated inflammatory diseases. Curr. Top. Microbiol. Immunol. 2010, 346, 279–299. [Google Scholar] [PubMed]
- Barber, D.F.; Bartolomé, A.; Hernandez, C.; Flores, J.M.; Redondo, C.; Fernandez-Arias, C.; Camps, M.; Rückle, T.; Schwarz, M.K.; Rodríguez, S.; et al. PI3Kγ inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat. Med. 2005, 11, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Chang, W.; Hurley, M.; Vignery, A.; Wu, D. Important roles of PI3Kγ in osteoclastogenesis and bone homeostasis. Proc. Natl. Acad. Sci. USA 2010, 107, 12901–12906. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, D.; Lembo, G. PI3Kγ in hypertension: A novel therapeutic target controlling vascular myogenic tone and target organ damage. Cardiovasc. Res. 2012, 95, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, D.; Vecchione, C.; Mascio, G.; Esposito, G.; Cifelli, G.; Martinello, K.; Landolfi, A.; Selvetella, G.; Grieco, P.; Damato, A.; et al. PI3Kγ inhibition reduces blood pressure by a vasorelaxant Akt/L-type calcium channel mechanism. Cardiovasc. Res. 2012, 93, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, C.; Patrucco, E.; Marino, G.; Barberis, L.; Poulet, R.; Aretini, A.; Maffei, A.; Gentile, M.T.; Storto, M.; Azzolino, O.; et al. Protection from angiotensin II-mediated vasculotoxic and hypertensive response in mice lacking PI3Kγ. J. Exp. Med. 2005, 201, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Penninger, J.M. Cardiac regulation by phosphoinositide 3-kinases and PTEN. Cardiovasc. Res. 2009, 82, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Crackower, M.A.; Oudit, G.Y.; Kozieradzki, I.; Sarao, R.; Sun, H.; Sasaki, T.; Hirsch, E.; Suzuki, A.; Shioi, T.; Irie-Sasaki, J.; et al. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell 2002, 110, 737–749. [Google Scholar] [CrossRef]
- Alloatti, G.; Montrucchio, G.; Lembo, G.; Hirsch, E. Phosphoinositide 3-kinase gamma: Kinase-dependent and -independent activities in cardiovascular function and disease. Biochem. Soc. Trans. 2004, 32, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Kassiri, Z.; Basu, R.; Chow, F.L.; Kandalam, V.; Damilano, F.; Liang, W.; Izumo, S.; Hirsch, E.; Penninger, J.M.; et al. Loss of PI3Kγ enhances cAMP-dependent MMP remodeling of the myocardial N-cadherin adhesion complexes and extracellular matrix in response to early biomechanical stress. Circ. Res. 2010, 107, 1275–1289. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Crackower, M.A.; Eriksson, U.; Sarao, R.; Kozieradzki, I.; Sasaki, T.; Irie-Sasaki, J.; Gidrewicz, D.; Rybin, V.O.; Wada, T.; et al. Phosphoinositide 3-kinase gamma-deficient mice are protected from isoproterenol-induced heart failure. Circulation 2003, 108, 2147–2152. [Google Scholar] [CrossRef] [PubMed]
- Damilano, F.; Franco, I.; Perrino, C.; Schaefer, K.; Azzolino, O.; Carnevale, D.; Cifelli, G.; Carullo, P.; Ragona, R.; Ghigo, A.; et al. Distinct effects of leukocyte and cardiac phosphoinositide 3-kinase γ activity in pressure overload-induced cardiac failure. Circulation 2011, 123, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Ghigo, A.; Ferrero, E.; Morello, F.; Santulli, G.; Baillie, G.S.; Damilano, F.; Dunlop, A.J.; Pawson, C.; Walser, R.; et al. Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110γ. Mol. Cell 2011, 42, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.D.; Sukhova, G.K.; Libby, P.; Schvartz, E.; Lichtenstein, A.H.; Field, S.J.; Kennedy, C.; Madhavarapu, S.; Luo, J.; Wu, D.; et al. Deletion of the phosphoinositide 3-kinase p110γ gene attenuates murine atherosclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 8077–8082. [Google Scholar] [CrossRef] [PubMed]
- Fougerat, A.; Gayral, S.; Gourdy, P.; Schambourg, A.; Rückle, T.; Schwarz, M.K.; Rommel, C.; Hirsch, E.; Arnal, J.F.; Salles, J.P.; et al. Genetic and pharmacological targeting of phosphoinositide 3-kinase-γ reduces atherosclerosis and favors plaque stability by modulating inflammatory processes. Circulation 2008, 117, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Maffei, A.; Cifelli, G.; Carnevale, R.; Iacobucci, R.; Pallante, F.; Fardella, V.; Fardella, S.; Hirsch, E.; Lembo, G.; Carnevale, D. PI3Kγ inhibition protects against diabetic cardiomyopathy in mice. Rev. Esp. Cardiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jiang, H.; Xie, W.; Zhang, Z.; Smrcka, A.V.; Wu, D. Roles of PLC-β2 and -β3 and PI3Kγ in chemoattractant-mediated signal transduction. Science 2000, 287, 1046–1049. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Irie-Sasaki, J.; Jones, R.G.; Oliveira-dos-Santos, A.J.; Stanford, W.L.; Bolon, B.; Wakeham, A.; Itie, A.; Bouchard, D.; Kozieradzki, I.; et al. Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science 2000, 287, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Nobs, S.P.; Schneider, C.; Dietrich, M.G.; Brocker, T.; Rolink, A.; Hirsch, E.; Kopf, M. PI3-Kinase-γ Has a Distinct and Essential Role in Lung-Specific Dendritic Cell Development. Immunity 2015, 43, 674–689. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.H.; Fraser, J.D.; Weiss, A. The cytoplasmic domain of CD28 is both necessary and sufficient for costimulation of interleukin-2 secretion and association with phosphatidylinositol 3-kinase. Mol. Cell. Biol. 1994, 14, 3392–3402. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.L.; Schwartz, M.D.; Jameson, S.C.; Shimizu, Y. Selective regulation of CD8 effector T cell migration by the p110γ isoform of phosphatidylinositol 3-kinase. J. Immunol. 2008, 180, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Ladygina, N.; Gottipati, S.; Ngo, K.; Castro, G.; Ma, J.Y.; Banie, H.; Rao, T.S.; Fung-Leung, W.P. PI3Kγ kinase activity is required for optimal T-cell activation and differentiation. Eur. J. Immunol. 2013, 43, 3183–3196. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Borlado, L.; Barber, D.F.; Hernández, C.; Rodríguez-Marcos, M.A.; Sánchez, A.; Hirsch, E.; Wymann, M.; Martínez-A, C.; Carrera, A.C. Phosphatidylinositol 3-kinase regulates the CD4/CD8 T cell differentiation ratio. J. Immunol. 2003, 170, 4475–4482. [Google Scholar] [CrossRef] [PubMed]
- Dutra, R.C.; Cola, M.; Leite, D.F.; Bento, A.F.; Claudino, R.F.; Nascimento, A.F.; Leal, P.C.; Calixto, J.B. Inhibitor of PI3Kγ ameliorates TNBS-induced colitis in mice by affecting the functional activity of CD4+CD25+FoxP3+ regulatory T cells. Br. J. Pharmacol. 2011, 163, 358–374. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lemus, L.A.; Crow, T.; Davis, M.J.; Meininger, G.A. αvβ3- and α5β1-integrin blockade inhibits myogenic constriction of skeletal muscle resistance arterioles. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H322–H329. [Google Scholar] [CrossRef] [PubMed]
- Sonoyama, K.; Greenstein, A.; Price, A.; Khavandi, K.; Heagerty, T. Vascular remodeling: Implications for small artery function and target organ damage. Ther. Adv. Cardiovasc. Dis. 2007, 1, 129–137. [Google Scholar] [CrossRef] [PubMed]
- House, S.J.; Potier, M.; Bisaillon, J.; Singer, H.A.; Trebak, M. The non-excitable smooth muscle: Calcium signaling and phenotypic switching during vascular disease. Pflüg. Arch. Eur. J. Physiol. 2008, 456, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995, 377, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.M.; Wang, G.R.; Lu, P.; Karas, R.H.; Aronovitz, M.; Heximer, S.P.; Kaltenbronn, K.M.; Blumer, K.J.; Siderovski, D.P.; Zhu, Y.; et al. Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat. Med. 2003, 9, 1506–1512. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Osol, G.; Brekke, J.F.; McElroy-Yaggy, K.; Gokina, N.I. Myogenic tone, reactivity, and forced dilatation: A three-phase model of in vitro arterial myogenic behavior. Am. J. Physiol. Heart Circ. Physiol. 2002, 2836, H2260–H2267. [Google Scholar] [CrossRef] [PubMed]
- Chlopicki, S.; Nilsson, H.; Mulvany, M.J. Initial and sustained phases of myogenic response of rat mesenteric small arteries. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H2176–H2183. [Google Scholar] [PubMed]
- Hill, M.A.; Meininger, G.A.; Davis, M.J.; Laher, I. Therapeutic potential of pharmacologically targeting arteriolar myogenic tone. Trends Pharmacol. Sci. 2009, 30, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Meininger, G.A.; Trzeciakowski, J.P. Vasoconstriction is amplified by autoregulation during vasoconstrictor-induced hypertension. Am. J. Physiol. Heart Circ. Physiol. 1988, 254, H709–H718. [Google Scholar]
- Hill, M.A.; Davis, M.J.; Meininger, G.A.; Potocnik, S.J.; Murphy, T.V. Arteriolar myogenic signalling mechanisms: Implications for local vascular function. Clin. Hemorheol. Microcirc. 2006, 34, 67–79. [Google Scholar] [PubMed]
- Le Blanc, C.; Mironneau, C.; Barbot, C.; Henaff, M.; Bondeva, T.; Wetzker, R.; Macrez, N. Regulation of vascular L-type Ca2+ channels by phosphatidylinositol 3,4,5-trisphosphate. Circ. Res. 2004, 95, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.F. PI3King the L-type calcium channel activation mechanism. Circ. Res. 2001, 89, 641–644. [Google Scholar] [PubMed]
- Carnevale, D.; Pallante, F.; Fardella, V.; Fardella, S.; Iacobucci, R.; Federici, M.; Cifelli, G.; de Lucia, M.; Lembo, G. The angiogenic factor PlGF mediates a neuroimmune interaction in the spleen to allow the onset of hypertension. Immunity 2014, 41, 737–752. [Google Scholar] [CrossRef] [PubMed]
- McMaster, W.G.; Kirabo, A.; Madhur, M.S.; Harrison, D.G. Inflammation, immunity, and hypertensive end-organ damage. Circ. Res. 2015, 116, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Johnson, G.V. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 2004, 29, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Zeiher, A.M. Nitric oxide-an endothelial cell survival factor. Cell Death Differ. 1999, 6, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Shiojima, I.; Walsh, K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ. Res. 2002, 90, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Rohrschneider, L.R.; Fuller, J.F.; Wolf, I.; Liu, Y.; Lucas, D.M. Structure, function, and biology of SHIP proteins. Genes Dev. 2000, 14, 505–520. [Google Scholar] [PubMed]
- Yamada, K.M.; Araki, M. Tumor suppressor PTEN: Modulator of cell signaling, growth, migration and apoptosis. J. Cell Sci. 2001, 114, 2375–2382. [Google Scholar] [PubMed]
- D’Andrea, I.; Fardella, V.; Fardella, S.; Pallante, F.; Ghigo, A.; Iacobucci, R.; Maffei, A.; Hirsch, E.; Lembo, G.; Carnevale, D. Lack of kinase-independent activity of PI3Kγ in locus coeruleus induces ADHD symptoms through increased CREB signaling. EMBO Mol. Med. 2015, 7, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Patrucco, E.; Notte, A.; Barberis, L.; Selvetella, G.; Maffei, A.; Brancaccio, M.; Marengo, S.; Russo, G.; Azzolino, O.; Rybalkin, S.D.; et al. PI3Kγ modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 2004, 118, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Quignard, J.F.; Mironneau, J.; Carricaburu, V.; Fournier, B.; Babich, A.; Nurnberg, B.; Mironneau, C.; Macrez, N. Phosphoinositide 3-kinase gamma mediates angiotensin II-induced stimulation of L-type calcium channels in vascular myocytes. J. Biol. Chem. 2001, 276, 32545–32551. [Google Scholar] [CrossRef] [PubMed]
- Wassmann, S.; Czech, T.; van Eickels, M.; Fleming, I.; Bohm, M.; Nickenig, G. Inhibition of diet-induced atherosclerosis and endothelial dysfunction in apolipoprotein E/angiotensin II type 1A receptor double-knockout mice. Circulation 2004, 110, 3062–3067. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.; Katanaev, V.L.; Garlanda, C.; Azzolino, O.; Pirola, L.; Silengo, L.; Sozzani, S.; Mantovani, A.; Altruda, F.; Wymann, M.P. Central role for G protein-coupled phosphoinositide 3-kinase γ in inflammation. Science 2000, 287, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Hilzendeger, A.M.; Cassell, M.D.; Davis, D.R.; Stauss, H.M.; Mark, A.L.; Grobe, J.L.; Sigmund, C.D. Angiotensin type 1A receptors in the subfornical organ are required for deoxycorticosterone acetate-salt hypertension. Hypertension 2013, 61, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Trott, D.W.; Thabet, S.R.; Kirabo, A.; Saleh, M.A.; Itani, H.; Norlander, A.E.; Wu, J.; Goldstein, A.; Arendshorst, W.J.; Madhur, M.S.; et al. Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension 2014, 64, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Vinh, A.; Chen, W.; Blinder, Y.; Weiss, D.; Taylor, W.R.; Goronzy, J.J.; Weyand, C.M.; Harrison, D.G.; Guzik, T.J. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation 2010, 122, 2529–2537. [Google Scholar] [CrossRef] [PubMed]
- Venable, J.D.; Ameriks, M.K.; Blevitt, J.M.; Thurmond, R.L.; FungLeung, W.P. Phosphoinositide 3-kinase γ (PI3Kγ)inhibitors for the treatment of inflammation and autoimmune disease. Recent Pat. Inflamm. Allergy Drug Discov. 2010, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Disease | Functions | References |
|---|---|---|
| Hypertension | L-type calcium channels in vascular myocytes | [15,16,17] |
| Heart Failure | Myocardial contractility; Cardiac remodeling | [18,19,20,21,22,23,24] |
| Atherosclerosis | Plaque stability | [25,26] |
| Diabetic Cardiomyopathy | Cardiac remodeling | [27] |
| Cell Type | Functions | References |
|---|---|---|
| Mast Cells | Hystamine release | [8,12] |
| Neutrophils | Inflammatory recruitment; chemoattractant-mediated signal transduction | [12,28] |
| Leukocytes | Inflammatory recruitment | [12] |
| Thymocyte | Thymocyte development | [29] |
| Myeloid cells | Osteoclastogenesis; bone homeostasis | [14] |
| Lung-Specific Dendritic Cell | Development | [30] |
| Lymphocytes | Inflammatory recruitment | [12] |
| B cell | T cell activation | [13] |
| T cell | Activation; migration; differentiation; CD4:CD8 T cells differentiation ratio | [31,32,33,34] |
| Treg | Activation | [35] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perrotta, M.; Lembo, G.; Carnevale, D. The Multifaceted Roles of PI3Kγ in Hypertension, Vascular Biology, and Inflammation. Int. J. Mol. Sci. 2016, 17, 1858. https://doi.org/10.3390/ijms17111858
Perrotta M, Lembo G, Carnevale D. The Multifaceted Roles of PI3Kγ in Hypertension, Vascular Biology, and Inflammation. International Journal of Molecular Sciences. 2016; 17(11):1858. https://doi.org/10.3390/ijms17111858
Chicago/Turabian StylePerrotta, Marialuisa, Giuseppe Lembo, and Daniela Carnevale. 2016. "The Multifaceted Roles of PI3Kγ in Hypertension, Vascular Biology, and Inflammation" International Journal of Molecular Sciences 17, no. 11: 1858. https://doi.org/10.3390/ijms17111858
APA StylePerrotta, M., Lembo, G., & Carnevale, D. (2016). The Multifaceted Roles of PI3Kγ in Hypertension, Vascular Biology, and Inflammation. International Journal of Molecular Sciences, 17(11), 1858. https://doi.org/10.3390/ijms17111858