
High Intra- and Inter-Tumoral Heterogeneity of RAS Mutations in Colorectal Cancer

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. Population
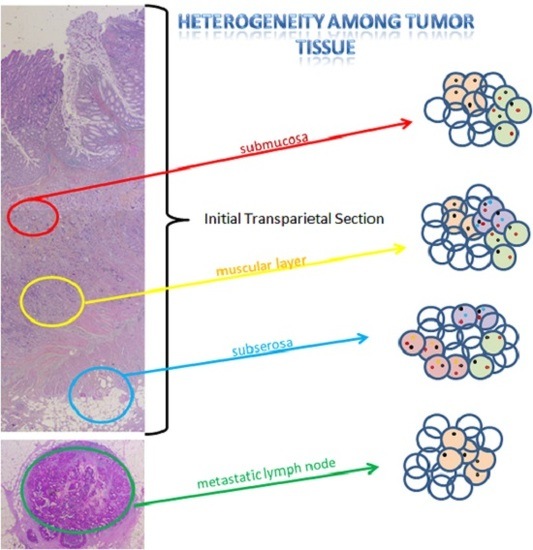
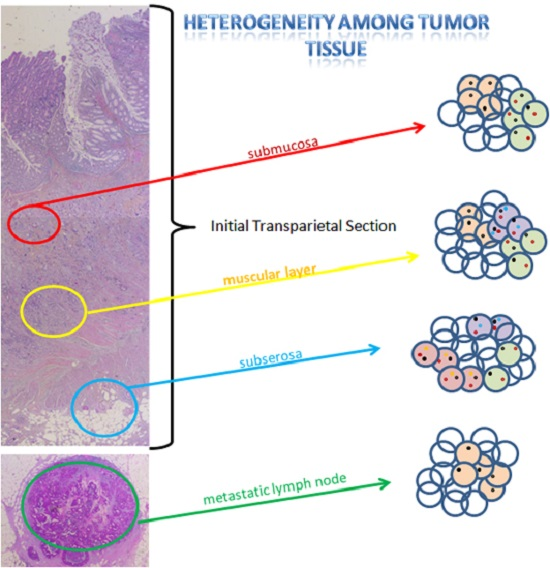
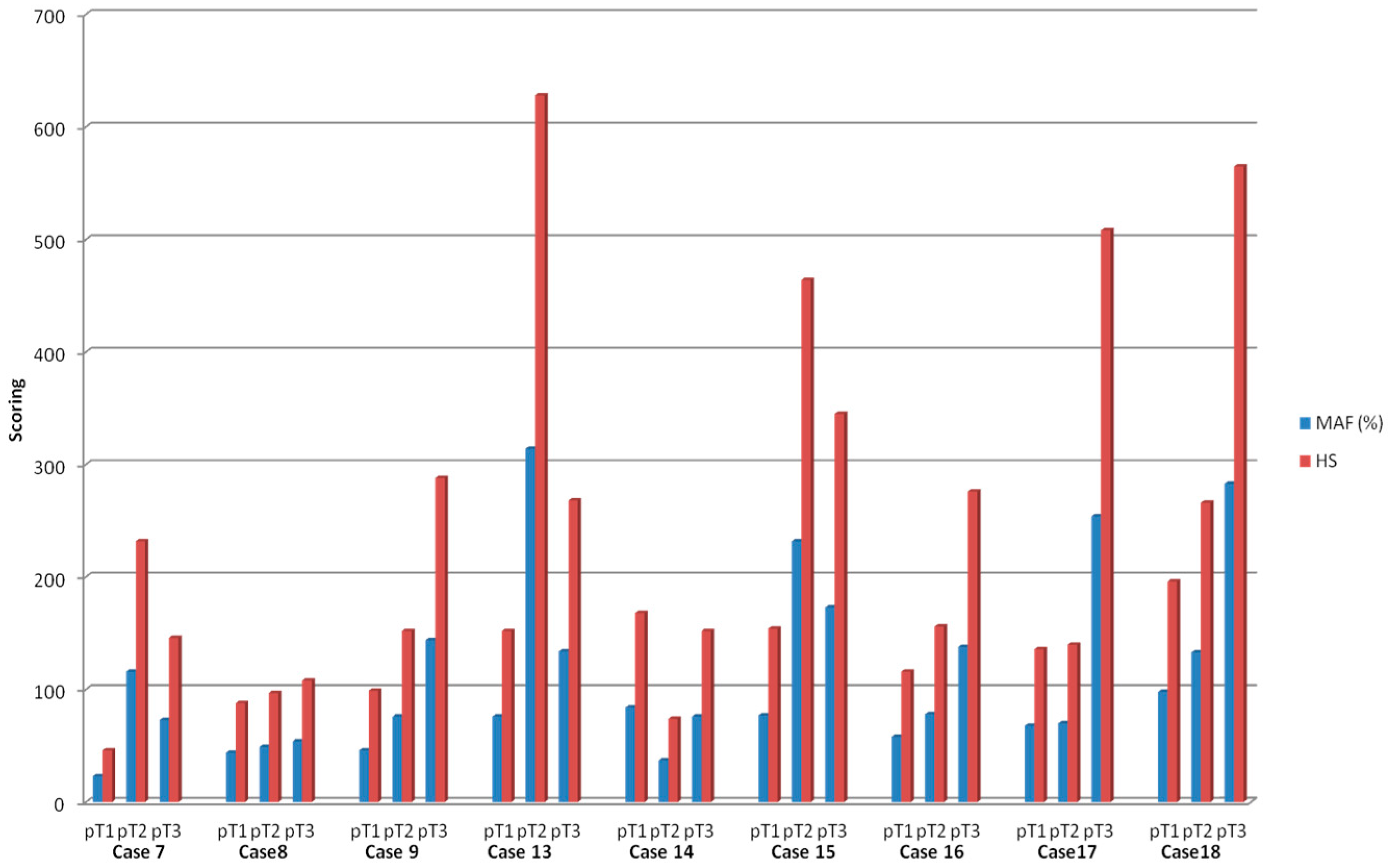
2.2. Intra-Tumoral and Inter-Tumoral Heterogeneity of KRAS and NRAS Mutations
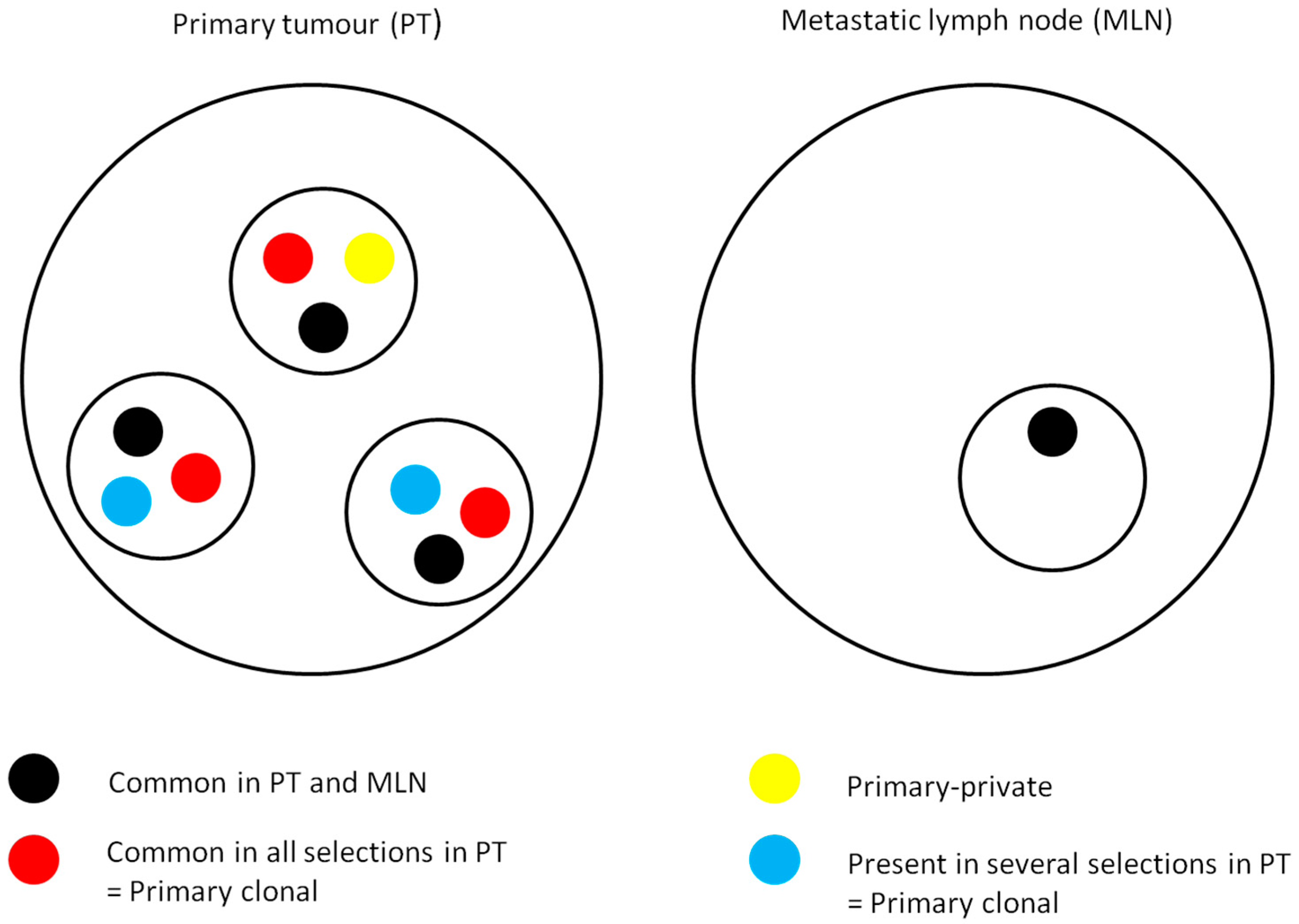
2.3. Mutational Intra-Tumoral Heterogeneity
2.4. RAS Mutations Are Not Exclusive
3. Discussion
4. Materials and Methods
4.1. Tumor Samples
4.2. KRAS, NRAS and BRAF Mutation Testing
4.3. Determination of Mutant Allele Burden and Tumoral Heterogeneity
4.4. Statistical Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Lenz, H.J.; Kohne, C.H.; Heinemann, V.; Tejpar, S.; Melezinek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.H.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Lievre, A.; Bachet, J.B.; le Corre, D.; Boige, V.; Landi, B.; Emile, J.F.; Cote, J.F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef] [PubMed]
- Sorich, M.J.; Wiese, M.D.; Rowland, A.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: A meta-analysis of randomized, controlled trials. Ann. Oncol. 2015, 26, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.; Dias, M.M.; Wiese, M.D.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S.; Sorich, M.J. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br. J. Cancer 2015, 112, 1888–1894. [Google Scholar] [CrossRef] [PubMed]
- Tougeron, D.; Lecomte, T.; Pages, J.C.; Villalva, C.; Collin, C.; Ferru, A.; Tourani, J.M.; Silvain, C.; Levillain, P.; Karayan-Tapon, L. Effect of low-frequency KRAS mutations on the response to anti-EGFR therapy in metastatic colorectal cancer. Ann. Oncol. 2013, 24, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Dunn, E.F.; Harari, P.M. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat. Rev. Clin. Oncol. 2010, 7, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Puig, P.; Pekin, D.; Normand, C.; Kotsopoulos, S.K.; Nizard, P.; Perez-Toralla, K.; Rowell, R.; Olson, J.; Srinivasan, P.; le Corre, D.; et al. Clinical relevance of KRAS-mutated subclones detected with picodroplet digital PCR in advanced colorectal cancer treated with anti-EGFR therapy. Clin. Cancer Res. 2015, 21, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Calvo, M.; Concha, A.; Figueroa, A.; Garrido, F.; Valladares-Ayerbes, M. Colorectal Cancer Classification and Cell Heterogeneity: A Systems Oncology Approach. Int. J. Mol. Sci. 2015, 16, 13610–13632. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Okami, J.; Kodama, K.; Higashiyama, M.; Kato, K. Intratumor heterogeneity of epidermal growth factor receptor mutations in lung cancer and its correlation to the response to gefitinib. Cancer Sci. 2008, 99, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Yancovitz, M.; Litterman, A.; Yoon, J.; Ng, E.; Shapiro, R.L.; Berman, R.S.; Pavlick, A.C.; Darvishian, F.; Christos, P.; Mazumdar, M.; et al. Intra- and inter-tumor heterogeneity of BRAFV600E mutations in primary and metastatic melanoma. PLoS ONE 2012, 7, e29336. [Google Scholar] [CrossRef] [PubMed]
- Katona, T.M.; Jones, T.D.; Wang, M.; Eble, J.N.; Billings, S.D.; Cheng, L. Genetically heterogeneous and clonally unrelated metastases may arise in patients with cutaneous melanoma. Am. J. Surg. Pathol. 2007, 31, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, H.; Bardelli, A.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B.; Velculescu, V.E. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 2002, 418, 934. [Google Scholar] [CrossRef] [PubMed]
- Brannon, A.R.; Vakiani, E.; Sylvester, B.E.; Scott, S.N.; McDermott, G.; Shah, R.H.; Kania, K.; Viale, A.; Oschwald, D.M.; Vacic, V.; et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014, 15, 454. [Google Scholar] [CrossRef] [PubMed]
- Santini, D.; Loupakis, F.; Vincenzi, B.; Floriani, I.; Stasi, I.; Canestrari, E.; Rulli, E.; Maltese, P.E.; Andreoni, F.; Masi, G.; et al. High concordance of KRAS status between primary colorectal tumors and related metastatic sites: Implications for clinical practice. Oncologist 2008, 13, 1270–1275. [Google Scholar] [CrossRef] [PubMed]
- Baldus, S.E.; Schaefer, K.L.; Engers, R.; Hartleb, D.; Stoecklein, N.H.; Gabbert, H.E. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin. Cancer Res. 2010, 16, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Rachiglio, A.M.; Lambiase, M.; Martinelli, E.; Fenizia, F.; Esposito, C.; Roma, C.; Troiani, T.; Rizzi, D.; Tatangelo, F.; et al. Heterogeneity of KRAS, NRAS, BRAF and PIK3CA mutations in metastatic colorectal cancer and potential effects on therapy in the CAPRI GOIM trial. Ann. Oncol. 2015, 26, 1710–1714. [Google Scholar] [CrossRef] [PubMed]
- Vagaja, N.N.; Parry, J.; McCallum, D.; Thomas, M.A.; Bentel, J.M. Are all RAS mutations the same? Coexisting KRAS and NRAS mutations in a caecal adenocarcinoma and contiguous tubulovillous adenoma. J. Clin. Pathol. 2015, 68, 657–660. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Kosmidou, V.; Oikonomou, E.; Vlassi, M.; Avlonitis, S.; Katseli, A.; Tsipras, I.; Mourtzoukou, D.; Kontogeorgos, G.; Zografos, G.; Pintzas, A. Tumor heterogeneity revealed by KRAS, BRAF, and PIK3CA pyrosequencing: KRAS and PIK3CA intratumor mutation profile differences and their therapeutic implications. Hum. Mutat. 2014, 35, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Improta, G.; Zupa, A.; Possidente, L.; Tartarone, A.; Pedicini, P.; Nappi, A.; Molinari, S.; Fraggetta, F.; Vita, G. Coexistence of two different mutations in codon 12 of the KRAS gene in colorectal cancer: Report of a case supporting the concept of tumoral heterogeneity. Oncol. Lett. 2013, 5, 1741–1743. [Google Scholar] [CrossRef] [PubMed]
- Baisse, B.; Bouzourene, H.; Saraga, E.P.; Bosman, F.T.; Benhattar, J. Intratumor genetic heterogeneity in advanced human colorectal adenocarcinoma. Int. J. Cancer 2001, 93, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Jung, S.H.; An, C.H.; Lee, S.H.; Baek, I.P.; Kim, M.S.; Park, S.W.; Rhee, J.K.; Lee, S.H.; Chung, Y.J. Subclonal Genomic Architectures of Primary and Metastatic Colorectal Cancer Based on Intratumoral Genetic Heterogeneity. Clin. Cancer Res. 2015, 21, 4461–4472. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Velho, S.; Moutinho, C.; Ferreira, A.; Preto, A.; Domingo, E.; Capelinha, A.F.; Duval, A.; Hamelin, R.; Machado, J.C.; et al. KRAS and BRAF oncogenic mutations in MSS colorectal carcinoma progression. Oncogene 2007, 26, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Richman, S.D.; Chambers, P.; Seymour, M.T.; Daly, C.; Grant, S.; Hemmings, G.; Quirke, P. Intra-tumoral heterogeneity of KRAS and BRAF mutation status in patients with advanced colorectal cancer (aCRC) and cost-effectiveness of multiple sample testing. Anal. Cell. Pathol. (Amst.) 2011, 34, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Molinari, F.; Felicioni, L.; Buscarino, M.; de Dosso, S.; Buttitta, F.; Malatesta, S.; Movilia, A.; Luoni, M.; Boldorini, R.; Alabiso, O.; et al. Increased detection sensitivity for KRAS mutations enhances the prediction of anti-EGFR monoclonal antibody resistance in metastatic colorectal cancer. Clin. Cancer Res. 2011, 17, 4901–4914. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Tougeron, D.; Cortes, U.; Ferru, A.; Villalva, C.; Silvain, C.; Tourani, J.M.; Levillain, P.; Karayan-Tapon, L. Epidermal growth factor receptor (EGFR) and KRAS mutations during chemotherapy plus anti-EGFR monoclonal antibody treatment in metastatic colorectal cancer. Cancer Chemother. Pharmacol. 2013, 72, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Paguirigan, A.L.; Smith, J.; Meshinchi, S.; Carroll, M.; Maley, C.; Radich, J.P. Single-cell genotyping demonstrates complex clonal diversity in acute myeloid leukemia. Sci. Transl. Med. 2015, 7, 281re2. [Google Scholar] [CrossRef] [PubMed]
- Gunderson, L.L.; Jessup, J.M.; Sargent, D.J.; Greene, F.L.; Stewart, A.K. Revised TN categorization for colon cancer based on national survival outcomes data. J. Clin. Oncol. 2010, 28, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Cortes, U.; Guilloteau, K.; Rouvreau, M.; Archaimbault, C.; Villalva, C.; Karayan-Tapon, L. Development of pyrosequencing methods for the rapid detection of RAS mutations in clinical samples. Exp. Mol. Pathol. 2015, 99, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Haley, L.; Tseng, L.H.; Zheng, G.; Dudley, J.; Anderson, D.A.; Azad, N.S.; Gocke, C.D.; Eshleman, J.R.; Lin, M.T. Performance characteristics of next-generation sequencing in clinical mutation detection of colorectal cancers. Mod. Pathol. 2015, 28, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
- Mohamed Suhaimi, N.-A.; Foong, Y.M.; Lee, D.Y.S.; Phyo, W.M.; Cima, I.; Lee, E.X.W.; Goh, W.L.; Lim, W.-Y.; Chia, K.S.; Kong, S.L.; et al. Non-invasive sensitive detection of KRAS and BRAF mutation in circulating tumor cells of colorectal cancer patients. Mol. Oncol. 2015, 9, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Basnet, S.; Zhang, Z.-Y.; Liao, W.-Q.; Li, S.-H.; Li, P.-S.; Ge, H.-Y. The Prognostic Value of Circulating Cell-Free DNA in Colorectal Cancer: A Meta-Analysis. J. Cancer 2016, 7, 1105–1113. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age | Sex | Tumor Site | Stage | pTNM 2009 | Initial KRAS | Initial BRAF | Recurrence | OS | Status |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 77.9 | F | right colon | III | pT3N1bM0 | WT | WT | yes | 48.68 | dead |
| 3 | 75.9 | M | left colon | III | pT4aN2bM0 | WT | WT | no | 2.50 | alive |
| 4 | 79.2 | F | right colon | II | pT3N0M0 | WT | V600E | no | 59.14 | alive |
| 6 | 55.9 | M | left colon | I | pT2N0M0 | WT | WT | no | 62.50 | alive |
| 7 | 67.3 | M | left colon | III | pT3N1bM0 | G12S | WT | yes | 69.21 | dead |
| 8 | 72.9 | F | left colon | IV | pT3N1bM1 | G12V | WT | - | 26.35 | dead |
| 9 | 58.5 | M | left colon | III | pT4aN0M0 | G12S | WT | yes | 82.86 | dead |
| 12 | 77.8 | F | left colon | IV | pT4aN0M1 | G12D | WT | - | 22.60 | dead |
| 14 | 56.4 | F | right colon | IV | pT3N1aM1 | G12V | WT | - | 48.39 | dead |
| 15 | 77.7 | M | left colon | IV | pT3N2bM1 | G12D | WT | - | 14.41 | dead |
| 16 | 74.7 | F | right colon | II | pT3N0M0 | G12V | WT | no | 37.70 | alive |
| 13 | 66.0 | M | left colon | II | pT3N0M0 | G12V | WT | - | 49.00 | alive |
| 1 | 61.9 | M | rectum | IV | pT3N2bM1 | WT | WT | - | 22.5 | dead |
| 5 | 58.9 | M | rectum | IV | pT3N0M1 | WT | WT | - | 52.34 | dead |
| 10 | 57.1 | F | rectum | IV | pT3N2bM1 | WT | WT | - | 44.80 | dead |
| 11 | 65.2 | F | rectum | IV | pT3N2bM1 | G12D | WT | - | 17.96 | dead |
| 17 | 54.9 | F | rectum | III | pT3N2aM0 | G12V | WT | yes | 41.22 | alive |
| 18 | 58.6 | M | rectum | IV | pT3N0M1 | G12R | WT | - | 43.82 | dead |
| Heterogeneity | Patients | pTNM | %TC | KRAS | Other Genes |
|---|---|---|---|---|---|
| Intra-tumoral heterogeneity | 4 | ITS | 70 | WT | BRAF:V600E (PLLM) |
| pT1 | 30 | WT | WT | ||
| pT2 | 90 | WT | BRAF:V600E (MUT) | ||
| 9 | ITS | 70 | G12S (PLLM)/Q61R (PLLM) | NRAS:Q61R (MUT) | |
| pT1 | 95 | WT | NRAS:Q61R (MUT) | ||
| pT2 | 70 | WT | NRAS:Q61R (MUT) | ||
| pT3 | 25 | WT | NRAS:Q61R (MUT) | ||
| 18 | ITS | 80 | G12R (MUT) | WT | |
| pT1 | 30 | G12R (MUT) | NRAS : K117N (PLLM) | ||
| pT2 | 35 | G12R (MUT) | WT | ||
| pT3 | 20 | G12R (MUT) | WT | ||
| Intra-tumoral and Inter-tumoral heterogeneity | 2 | ITS | 80 | WT | NRAS: A59T (PLLM) |
| pT2 | 15 | WT | WT | ||
| pT3 | 20 | G13D (MUT) | WT | ||
| N | 5 | WT | WT | ||
| 7 | ITS | 60 | G12S (PLLM) | FA (NRAS61 et KRAS146) | |
| pT1 | 70 | G12D (MUT)/Q61L (PLLM) | NRAS:Q61K (MUT) | ||
| pT2 | 25 | G12S (MUT) | NRAS:Q61K (MUT) | ||
| pT3 | 40 | Q61L (PLLM) | NRAS:Q61K (MUT) | ||
| N | 25 | WT | NRAS:Q61K (MUT) | ||
| 12 | ITS | 40 | G12D (PLLM)/A146T (MUT) | WT | |
| pT2 | 15 | A146T (MUT) | WT | ||
| pT3 | 20 | Q61H(PLLM)/A146T (MUT) | FA (BRAF) | ||
| M | 25 | WT | FA(NRAS59) | ||
| Inter-tumoral heterogeneity only | 14 | ITS | 30 | G12V (MUT) | WT |
| pT1 | 5 | G12V (PLLM) | WT | ||
| pT2 | 40 | G12V (MUT) | WT | ||
| pT3 | 5 | G12V (PLLM) | WT | ||
| N | 5 | WT | WT | ||
| Mutation without heterogeneity | 8 | ITS | 30 | G12V (MUT) | WT |
| pT1 | 60 | G12V (MUT) | WT | ||
| pT2 | 75 | G12V (MUT) | WT | ||
| pT3 | 60 | G12V (MUT) | WT | ||
| N | 80 | G12V (MUT) | WT | ||
| 11 | ITS | 5 | G12D (PLLM) | WT | |
| pT2 | 5 | G12D (MUT) | WT | ||
| pT3 | 5 | G12D (PLLM) | WT | ||
| N | 5 | G12D (MUT) | WT | ||
| 13 | ITS | 70 | G12V (MUT) | WT | |
| pT1 | 70 | G12V (MUT) | WT | ||
| pT2 | 10 | G12V (MUT) | WT | ||
| pT3 | 15 | G12V (MUT) | WT | ||
| 15 | ITS | 40 | G12D (MUT) | WT | |
| pT1 | 60 | G12D (MUT) | WT | ||
| pT2 | 15 | G12D (MUT) | WT | ||
| pT3 | 20 | G12D (MUT) | WT | ||
| N | 40 | G12D (MUT) | WT | ||
| 16 | ITS | 40 | G12V (MUT) | WT | |
| pT1 | 80 | G12V (MUT) | WT | ||
| pT2 | 40 | G12V (MUT) | WT | ||
| pT3 | 10 | G12V (MUT) | WT | ||
| 17 | ITS | 30 | G12V (MUT) | WT | |
| pT1 | 30 | G12V (MUT) | WT | ||
| pT2 | 20 | G12V (MUT) | WT | ||
| pT3 | 10 | G12V (MUT) | WT | ||
| N | 20 | G12V (MUT) | WT |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeantet, M.; Tougeron, D.; Tachon, G.; Cortes, U.; Archambaut, C.; Fromont, G.; Karayan-Tapon, L. High Intra- and Inter-Tumoral Heterogeneity of RAS Mutations in Colorectal Cancer. Int. J. Mol. Sci. 2016, 17, 2015. https://doi.org/10.3390/ijms17122015
Jeantet M, Tougeron D, Tachon G, Cortes U, Archambaut C, Fromont G, Karayan-Tapon L. High Intra- and Inter-Tumoral Heterogeneity of RAS Mutations in Colorectal Cancer. International Journal of Molecular Sciences. 2016; 17(12):2015. https://doi.org/10.3390/ijms17122015
Chicago/Turabian StyleJeantet, Marion, David Tougeron, Gaelle Tachon, Ulrich Cortes, Céline Archambaut, Gaelle Fromont, and Lucie Karayan-Tapon. 2016. "High Intra- and Inter-Tumoral Heterogeneity of RAS Mutations in Colorectal Cancer" International Journal of Molecular Sciences 17, no. 12: 2015. https://doi.org/10.3390/ijms17122015
APA StyleJeantet, M., Tougeron, D., Tachon, G., Cortes, U., Archambaut, C., Fromont, G., & Karayan-Tapon, L. (2016). High Intra- and Inter-Tumoral Heterogeneity of RAS Mutations in Colorectal Cancer. International Journal of Molecular Sciences, 17(12), 2015. https://doi.org/10.3390/ijms17122015