
Using CRISPR/Cas9-Mediated GLA Gene Knockout as an In Vitro Drug Screening Model for Fabry Disease

 , ,
, , 

Abstract
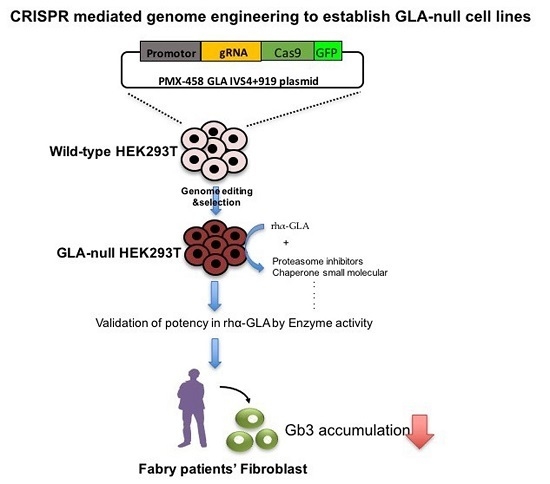
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
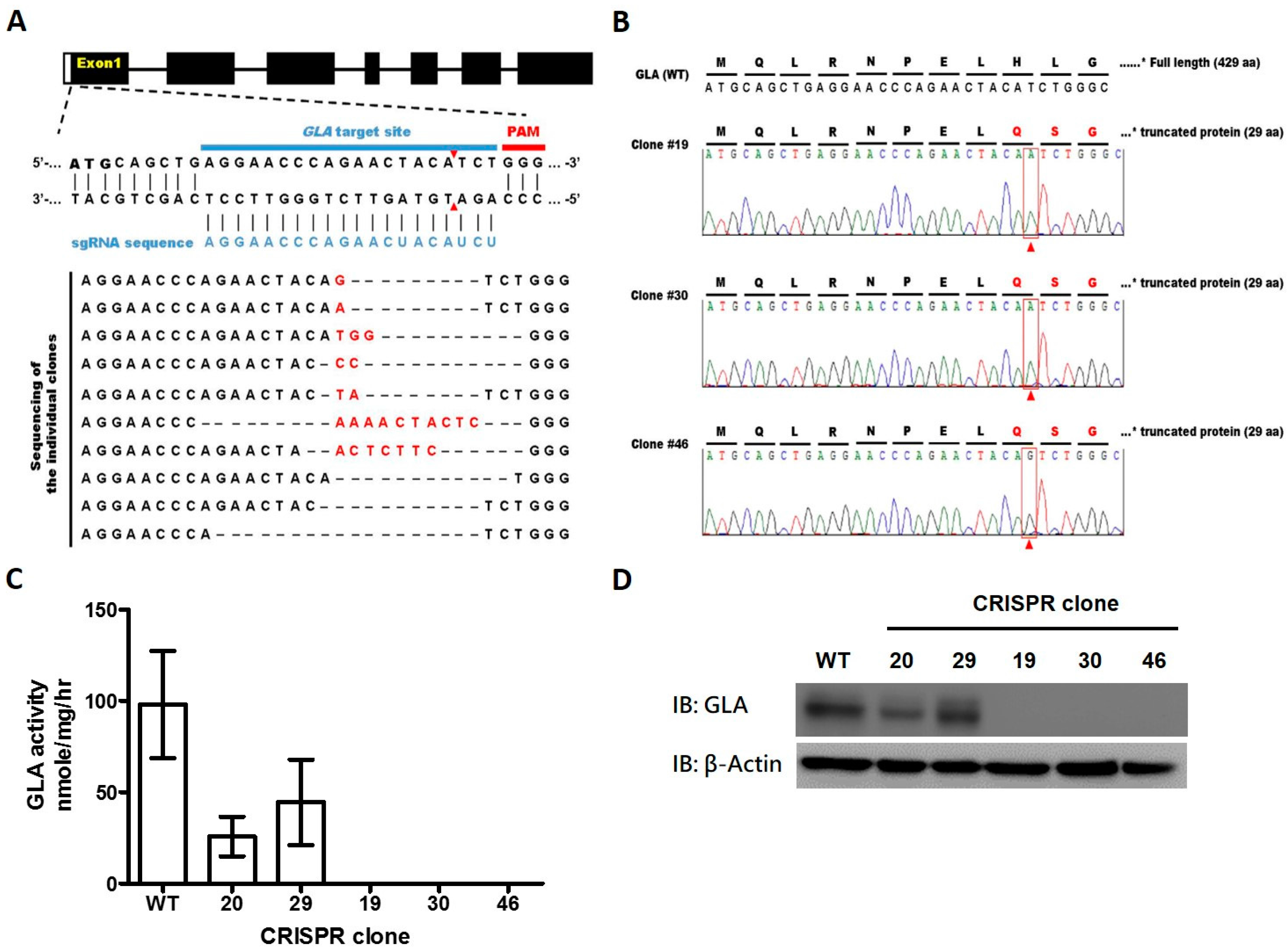
2.1. CRISPR/Cas9-Mediated Gene Editing of GLA Effectively and Completely Ablated Endogenous GLA Protein Expression in Human Cells
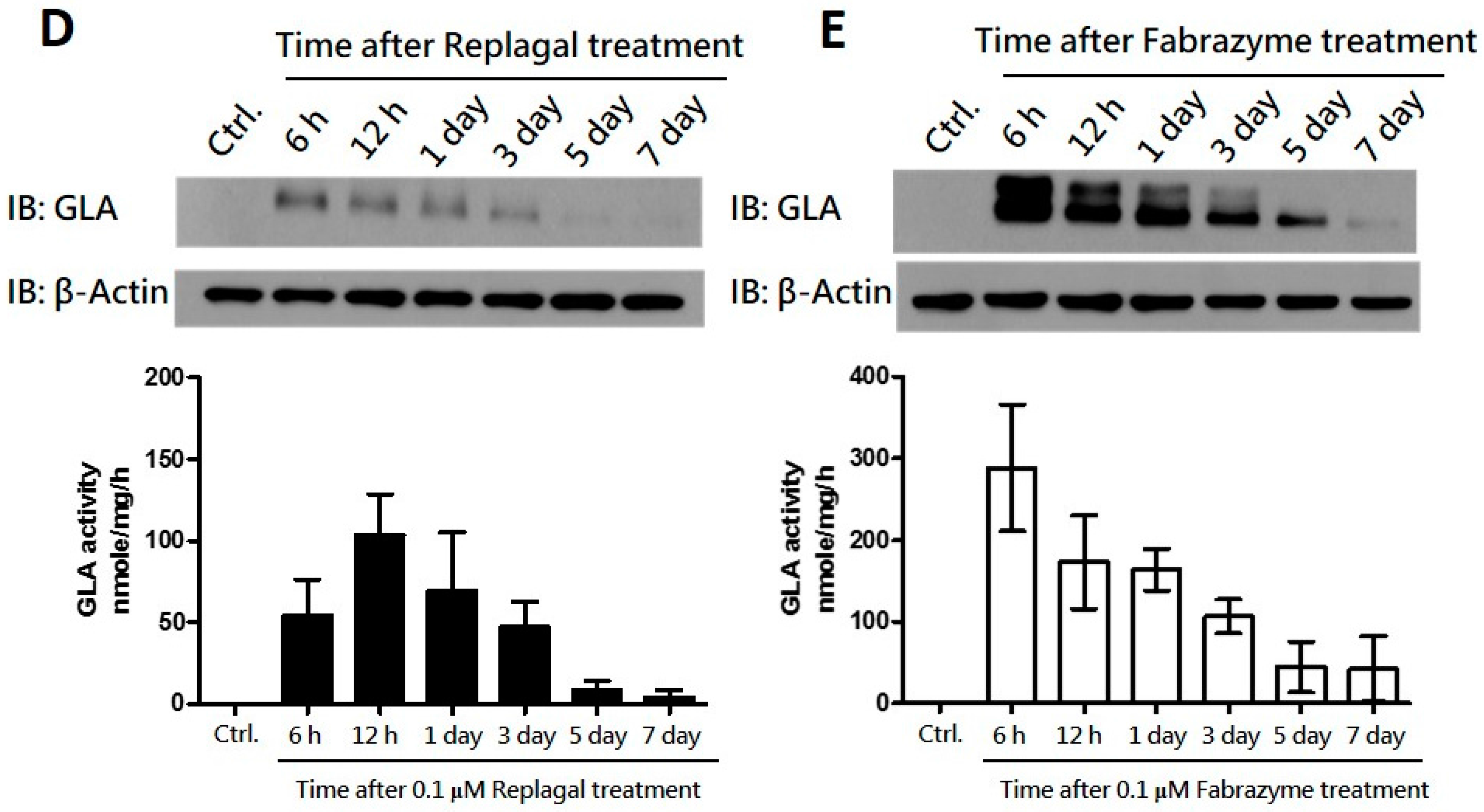
2.2. HEK-293T GLA-Null Cell Lines Serve as a Platform for rhα-GLA Intracellular Pharmacokinetics Assay
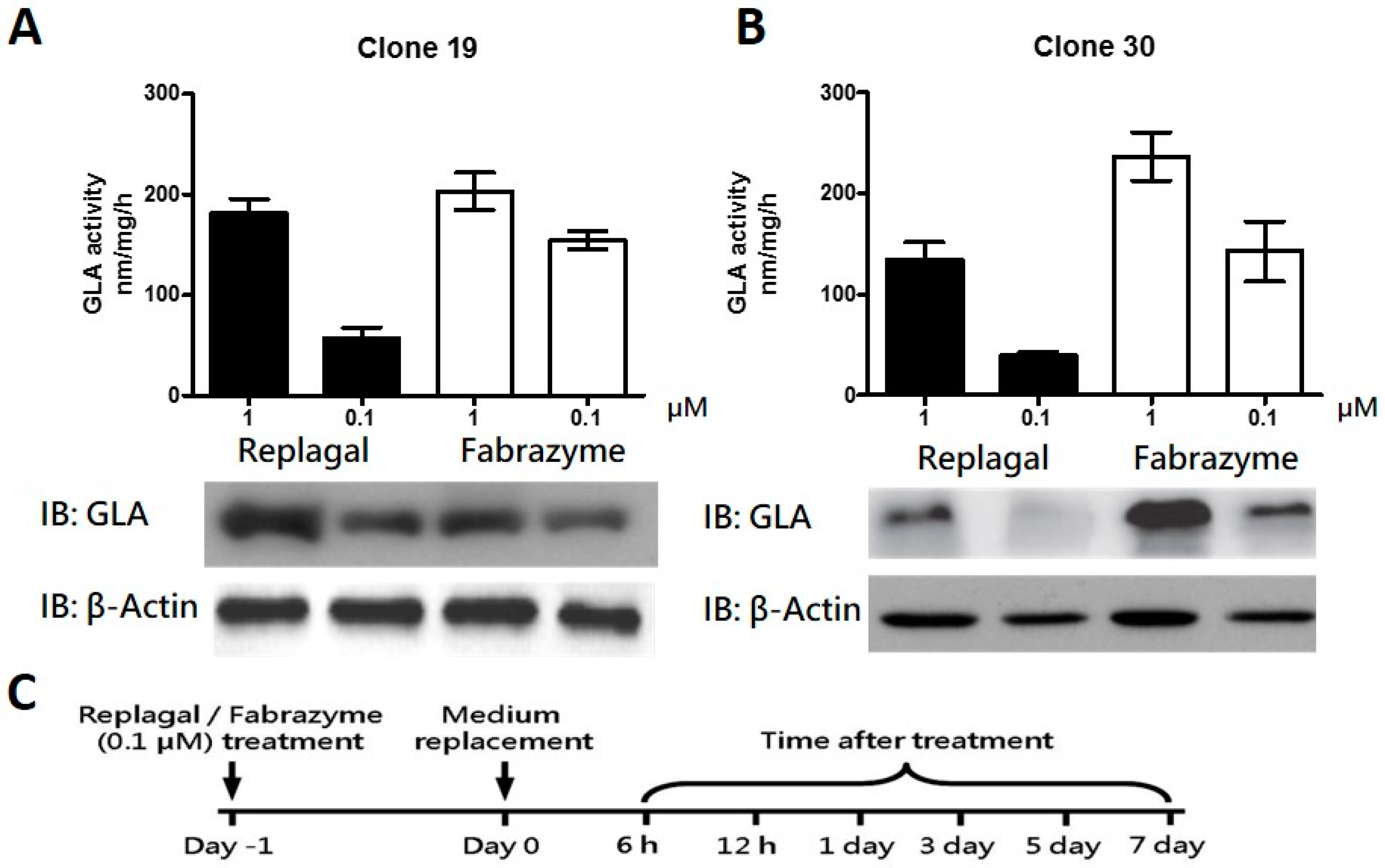
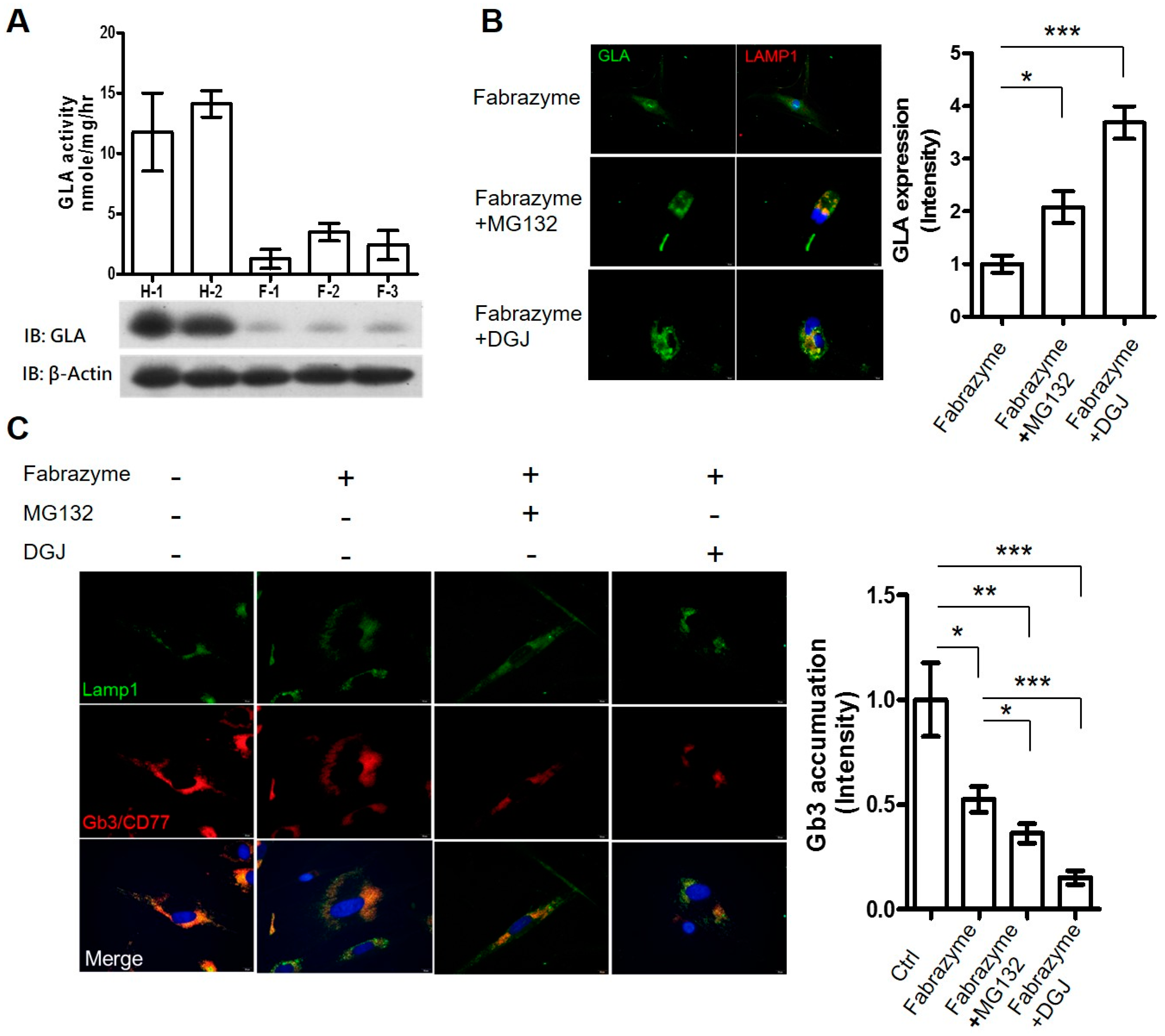
2.3. Co-Administration of MG132 Improved the Stability and Activity of rhα-GLA in the GLA-Null Cell Line without Cytotoxic Effects
2.4. MG132 Maintained Intracellular Amount of rhα-GLA and Reduced Gb3 Accumulation in Fabry Patient-Derived Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. CRISPR/Cas9 Plasmid Construction and Transfection
4.3. Analysis of CRISPR/Cas9-Mediated Indel Formation in GLA Gene
4.4. Immunofluorescence Stain
4.5. Western Blotting Analysis
4.6. GLA Enzyme Activity
4.7. Cell Viability Assay
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| sgRNA | single-guide RNA |
| Cas9 | CRISPR-associated protein |
| DGJ | 1-deoxygalactonojirimycin |
| ERT | enzyme replacement therapy |
| FD | Fabry disease |
| Gb3 | globotriaosylceramide |
| α-Gal A | α-galactosidase A |
| KO | knockout |
| HEK-293T | human embryonic kidney 293T cells |
| HSP | heat shock protein |
| LSD | lysosomal storage disease |
| PC | pharmacological chaperone |
| rhα-GLA | recombinant human α Gal A |
References
- Tsakiris, D.; Simpson, H.K.; Jones, E.H.; Briggs, J.D.; Elinder, C.G.; Mendel, S.; Piccoli, G.; dos Santos, J.P.; Tognoni, G.; Vanrenterghem, Y.; et al. Report on management of renale failure in Europe, XXVI, 1995. Rare diseases in renal replacement therapy in the ERA-EDTA Registry. Nephrol. Dial. Transplant. 1996, 11, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Desnick, R.J.; Wasserstein, M.P.; Banikazemi, M. Fabry disease (α-galactosidase A deficiency): Renal involvement and enzyme replacement therapy. Contrib. Nephrol. 2001, 136, 174–192. [Google Scholar]
- Hopkin, R.J.; Bissler, J.; Banikazemi, M.; Clarke, L.; Eng, C.M.; Germain, D.P.; Lemay, R.; Tylki-Szymanska, A.; Wilcox, W.R. Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. Pediatr. Res. 2008, 64, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.M.; Banikazemi, M.; Gordon, R.E.; Goldman, M.; Phelps, R.; Kim, L.; Gass, A.; Winston, J.; Dikman, S.; Fallon, J.T.; et al. A phase 1/2 clinical trial of enzyme replacement in fabry disease: Pharmacokinetic, substrate clearance, and safety studies. Am. J. Hum. Genet. 2001, 68, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Kopp, J.B.; Austin, H.A., 3rd; Sabnis, S.; Moore, D.F.; Weibel, T.; Balow, J.E.; Brady, R.O. Enzyme replacement therapy in Fabry disease: A randomized controlled trial. JAMA 2001, 285, 2743–2749. [Google Scholar] [CrossRef] [PubMed]
- Simoens, S. Pricing and reimbursement of orphan drugs: The need for more transparency. Orphanet J. Rare Dis. 2011, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Benichou, B.; Goyal, S.; Sung, C.; Norfleet, A.M.; O’Brien, F. A retrospective analysis of the potential impact of IgG antibodies to agalsidase β on efficacy during enzyme replacement therapy for Fabry disease. Mol. Genet. Metab. 2009, 96, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S. Pharmacological chaperone therapy for Fabry disease. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y. Chaperone therapy update: Fabry disease, GM1-gangliosidosis and Gaucher disease. Brain Dev. 2013, 35, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Valenzano, K.J.; Khanna, R.; Powe, A.C.; Boyd, R.; Lee, G.; Flanagan, J.J.; Benjamin, E.R. Identification and characterization of pharmacological chaperones to correct enzyme deficiencies in lysosomal storage disorders. Assay Drug Dev. Technol. 2011, 9, 213–235. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Chang, H.H.; Kawasaki, K.; Yasuda, K.; Wu, H.L.; Garman, S.C.; Fan, J.Q. Mutant α-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: Biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. Biochem. J. 2007, 406, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, G.H.; Kim, S.S.; Ko, J.M.; Lee, J.J.; Yoo, H.W. Effects of a chemical chaperone on genetic mutations in α-galactosidase A in Korean patients with Fabry disease. Exp. Mol. Med. 2009, 41, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Mena-Barragan, T.; Higaki, K.; Johnson, J.L.; Drury, J.E.; Lieberman, R.L.; Nakasone, N.; Ninomiya, H.; Tsukimura, T.; Sakuraba, H.; et al. Molecular basis of 1-deoxygalactonojirimycin arylthiourea binding to human α-galactosidase A: Pharmacological chaperoning efficacy on Fabry disease mutants. ACS Chem. Biol. 2014, 9, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.Q.; Ishii, S.; Asano, N.; Suzuki, Y. Accelerated transport and maturation of lysosomal α-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat. Med. 1999, 5, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.; Shinohara, T.; Yano, S.; Nakamura, M.; Yasuda, A.; Yokoyama, S.; Fan, J.Q.; Kawasaki, K.; Watanabe, M.; Ishii, S. Rescue of mutant α-galactosidase A in the endoplasmic reticulum by 1-deoxygalactonojirimycin leads to trafficking to lysosomes. Biochim. Biophys. Acta 2008, 1782, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, K.; Tajima, Y.; Kawashima, I.; Tsukimura, T.; Saito, S.; Ohno, K.; Iwamoto, K.; Kobayashi, T.; Itoh, K.; Sakuraba, H. Molecular interaction of imino sugars with human α-galactosidase: Insight into the mechanism of complex formation and pharmacological chaperone action in Fabry disease. Mol. Genet. Metab. 2009, 96, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.R.; Khanna, R.; Schilling, A.; Flanagan, J.J.; Pellegrino, L.J.; Brignol, N.; Lun, Y.; Guillen, D.; Ranes, B.E.; Frascella, M.; et al. Co-administration with the pharmacological chaperone AT1001 increases recombinant human α-galactosidase A tissue uptake and improves substrate reduction in Fabry mice. Mol. Ther. 2012, 20, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Lun, Y.; Brignol, N.; Hamler, R.; Schilling, A.; Frascella, M.; Sullivan, S.; Boyd, R.E.; Chang, K.; Soska, R.; et al. Coformulation of a novel human α-galactosidase a with the pharmacological chaperone AT1001 leads to improved substrate reduction in fabry mice. Mol. Ther. 2015, 23, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, T.; Murray, G.J.; Swaim, W.D.; Longenecker, G.; Quirk, J.M.; Cardarelli, C.O.; Sugimoto, Y.; Pastan, I.; Gottesman, M.M.; Brady, R.O.; et al. α-Galactosidase A deficient mice: A model of Fabry disease. Proc. Natl. Acad. Sci. USA 1997, 94, 2540–2544. [Google Scholar] [CrossRef] [PubMed]
- Porto, C.; Pisani, A.; Rosa, M.; Acampora, E.; Avolio, V.; Tuzzi, M.R.; Visciano, B.; Gagliardo, C.; Materazzi, S.; la Marca, G.; et al. Synergy between the pharmacological chaperone 1-deoxygalactonojirimycin and the human recombinant α-galactosidase A in cultured fibroblasts from patients with Fabry disease. J. Inherit. Metab. Dis. 2012, 35, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Jin, X.; Zhang, K.; Copertino, L.; Andrews, L.; Baker-Malcolm, J.; Geagan, L.; Qiu, H.; Seiger, K.; Barngrover, D.; et al. A biochemical and pharmacological comparison of enzyme replacement therapies for the glycolipid storage disorder Fabry disease. Glycobiology 2003, 13, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, M.; Brakch, N.; Celine, L.; Genty, D.; Ramdani, Y.; Moll, S.; Djavaheri-Mergny, M.; Brasse-Lagnel, C.; Annie Laquerriere, A.L.; Barbey, F.; et al. Autophagosome maturation is impaired in Fabry disease. Autophagy 2010, 6, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Almond, J.B.; Cohen, G.M. Cohen, The proteasome: A novel target for cancer chemotherapy. Leukemia 2002, 16, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chong, K.W.; Hsu, J.H.; Yu, H.C.; Shih, C.C.; Huang, C.H.; Lin, S.J.; Chen, C.H.; Chiang, C.C.; Ho, H.J.; et al. High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ. Cardiovasc. Genet. 2009, 2, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Hwu, W.L.; Chien, Y.H.; Lee, N.C.; Chiang, S.C.; Dobrovolny, R.; Huang, A.C.; Yeh, H.Y.; Chao, M.C.; Lin, S.J.; Kitagawa, T.; et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A). Hum. Mutat. 2009, 30, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Lee, N.C.; Chiang, S.C.; Desnick, R.J.; Hwu, W.L. Fabry disease: Incidence of the common later-onset α-galactosidase A IVS4+919G→A mutation in Taiwanese newborns—Superiority of DNA-based to enzyme-based newborn screening for common mutations. Mol. Med. 2012, 18, 780–784. [Google Scholar] [CrossRef] [PubMed]
- Seah, Y.F.; El Farran, C.A.; Warrier, T.; Xu, J.; Loh, Y.H. Induced pluripotency and gene editing in disease modelling: Perspectives and challenges. Int. J. Mol. Sci. 2015, 16, 28614–28634. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Rottbauer, W.; Just, S. Recent progress in the use of zebrafish for novel cardiac drug discovery. Expert Opin. Drug Discov. 2015, 10, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.M.; Labilloy, A.; Eshbach, M.L.; Roy, A.; Subramanya, A.R.; Monte, S.; Labilloy, G.; Weisz, O.A. Characterization and phosphoproteomic analysis of a human immortalized podocyte model of Fabry disease generated using CRISPR/Cas9 technology. Am. J. Physiol. Ren. Physiol. 2016, 311, F1015–F1024. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Katz, E.; Della Valle, M.C.; Mascioli, K.; Flanagan, J.J.; Castelli, J.P.; Schiffmann, R.; Boudes, P.; Lockhart, D.J.; Valenzano, K.J.; et al. A pharmacogenetic approach to identify mutant forms of α-galactosidase A that respond to a pharmacological chaperone for Fabry disease. Hum. Mutat. 2011, 32, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Giugliani, R.; Hughes, D.A.; Mehta, A.; Nicholls, K.; Barisoni, L.; Jennette, C.J.; Bragat, A.; Castelli, J.; Sitaraman, S.; et al. Safety and pharmacodynamic effects of a pharmacological chaperone on α-galactosidase A activity and globotriaosylceramide clearance in Fabry disease: Report from two phase 2 clinical studies. Orphanet J. Rare Dis. 2012, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Waldek, S.; Germain, D.P.; Nicholls, K.; Bichet, D.G.; Simosky, J.K.; Bragat, A.C.; Castelli, J.P.; Benjamin, E.R.; Boudes, P.F. A Phase 2 study of migalastat hydrochloride in females with Fabry disease: Selection of population, safety and pharmacodynamic effects. Mol. Genet. Metab. 2013, 109, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Hopkin, R.J.; Bissler, J.; Grabowski, G.A. Comparative evaluation of α-galactosidase A infusions for treatment of Fabry disease. Genet. Med. 2003, 5, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Pisani, A.; Visciano, B.; Roux, G.D.; Sabbatini, M.; Porto, C.; Parenti, G.; Imbriaco, M. Enzyme replacement therapy in patients with Fabry disease: State of the art and review of the literature. Mol. Genet. Metab. 2012, 107, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Lidove, O.; West, M.L.; Pintos-Morell, G.; Reisin, R.; Nicholls, K.; Figuera, L.E.; Parini, R.; Carvalho, L.R.; Kampmann, C.; Pastores, G.M.; et al. Effects of enzyme replacement therapy in Fabry disease—A comprehensive review of the medical literature. Genet. Med. 2010, 12, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Rombach, S.M.; Smid, B.E.; Bouwman, M.G.; Linthorst, G.E.; Dijkgraaf, M.G.; Hollak, C.E. Long term enzyme replacement therapy for Fabry disease: Effectiveness on kidney, heart and brain. Orphanet J. Rare Dis. 2013, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Charrow, J.; Desnick, R.J.; Guffon, N.; Kempf, J.; Lachmann, R.H.; Lemay, R.; Linthorst, G.E.; Packman, S.; Scott, C.R.; et al. Ten-year outcome of enzyme replacement therapy with agalsidase β in patients with Fabry disease. J. Med. Genet. 2015, 52, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Khanna, R.; Soska, R.; Lun, Y.; Feng, J.; Frascella, M.; Young, B.; Brignol, N.; Pellegrino, L.; Sitaraman, S.A.; Desnick, R.J.; et al. The pharmacological chaperone 1-deoxygalactonojirimycin reduces tissue globotriaosylceramide levels in a mouse model of Fabry disease. Mol. Ther. 2010, 18, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; Citro, V.; de Crescenzo, A.; Orlando, P.; Cammisa, M.; Correra, A.; Cubellis, M.V. Therapy of Fabry disease with pharmacological chaperones: From in silico predictions to in vitro tests. Orphanet J. Rare Dis. 2011, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, S.; Bembi, B.; Rosso, N.; Filocamo, M.; Dardis, A. Treatment of human fibroblasts carrying NPC1 missense mutations with MG132 leads to an improvement of intracellular cholesterol trafficking. JIMD Rep. 2012, 2, 59–69. [Google Scholar] [PubMed]
- Shimada, Y.; Nishida, H.; Nishiyama, Y.; Kobayashi, H.; Higuchi, T.; Eto, Y.; Ida, H.; Ohashi, T. Proteasome inhibitors improve the function of mutant lysosomal α-glucosidase in fibroblasts from Pompe disease patient carrying c.546G>T mutation. Biochem. Biophys. Res. Commun. 2011, 415, 274–278. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, E.M.; Igdoura, S.A. The therapeutic potential of pharmacological chaperones and proteosomal inhibitors, Celastrol and MG132 in the treatment of sialidosis. Mol. Genet. Metab. 2012, 107, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Pockrandt, A.M.; Seemann, S.; Sharif, M.; Runge, F.; Pohlers, S.; Zheng, C.; Glaser, A.; Beller, M.; Rolfs, A.; et al. Enzyme enhancers for the treatment of Fabry and Pompe disease. Mol. Ther. 2015, 23, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Stangl, K.; Gunther, C.; Frank, T.; Lorenz, M.; Meiners, S.; Ropke, T.; Stelter, L.; Moobed, M.; Baumann, G.; Kloetzel, P.M.; et al. Inhibition of the ubiquitin-proteasome pathway induces differential heat-shock protein response in cardiomyocytes and renders early cardiac protection. Biochem. Biophys. Res. Commun. 2002, 291, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, N.; Wagner, B.J. Upregulation of heat shock protein expression by proteasome inhibition: An antiapoptotic mechanism in the lens. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2082–2091. [Google Scholar] [CrossRef] [PubMed]
- Mu, T.W.; Ong, D.S.; Wang, Y.J.; Balch, W.E.; Yates, J.R., 3rd; Segatori, L.; Kelly, J.W. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell 2008, 134, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T.; Roth, A.G.; Petersen, N.H.; Mahalka, A.K.; Olsen, O.D.; Moilanen, I.; Zylicz, A.; Knudsen, J.; Sandhoff, K.; Arenz, C.; et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 2010, 463, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Ingemann, L.; Kirkegaard, T. Lysosomal storage diseases and the heat shock response: Convergences and therapeutic opportunities. J. Lipid Res. 2014, 55, 2198–2210. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR/Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, H.-Y.; Chiang, H.-C.; Tseng, W.-L.; Wu, P.; Chien, C.-S.; Leu, H.-B.; Yang, Y.-P.; Wang, M.-L.; Jong, Y.-J.; Chen, C.-H.; et al. Using CRISPR/Cas9-Mediated GLA Gene Knockout as an In Vitro Drug Screening Model for Fabry Disease. Int. J. Mol. Sci. 2016, 17, 2089. https://doi.org/10.3390/ijms17122089
Song H-Y, Chiang H-C, Tseng W-L, Wu P, Chien C-S, Leu H-B, Yang Y-P, Wang M-L, Jong Y-J, Chen C-H, et al. Using CRISPR/Cas9-Mediated GLA Gene Knockout as an In Vitro Drug Screening Model for Fabry Disease. International Journal of Molecular Sciences. 2016; 17(12):2089. https://doi.org/10.3390/ijms17122089
Chicago/Turabian StyleSong, Hui-Yung, Huai-Chih Chiang, Wei-Lien Tseng, Ping Wu, Chian-Shiu Chien, Hsin-Bang Leu, Yi-Ping Yang, Mong-Lien Wang, Yuh-Jyh Jong, Chung-Hsuan Chen, and et al. 2016. "Using CRISPR/Cas9-Mediated GLA Gene Knockout as an In Vitro Drug Screening Model for Fabry Disease" International Journal of Molecular Sciences 17, no. 12: 2089. https://doi.org/10.3390/ijms17122089
APA StyleSong, H. -Y., Chiang, H. -C., Tseng, W. -L., Wu, P., Chien, C. -S., Leu, H. -B., Yang, Y. -P., Wang, M. -L., Jong, Y. -J., Chen, C. -H., Yu, W. -C., & Chiou, S. -H. (2016). Using CRISPR/Cas9-Mediated GLA Gene Knockout as an In Vitro Drug Screening Model for Fabry Disease. International Journal of Molecular Sciences, 17(12), 2089. https://doi.org/10.3390/ijms17122089