
Synthesis, Bioevaluation and Molecular Dynamic Simulation Studies of Dexibuprofen–Antioxidant Mutual Prodrugs

 and
and
Abstract
:1. Introduction
2. Result and Discussion
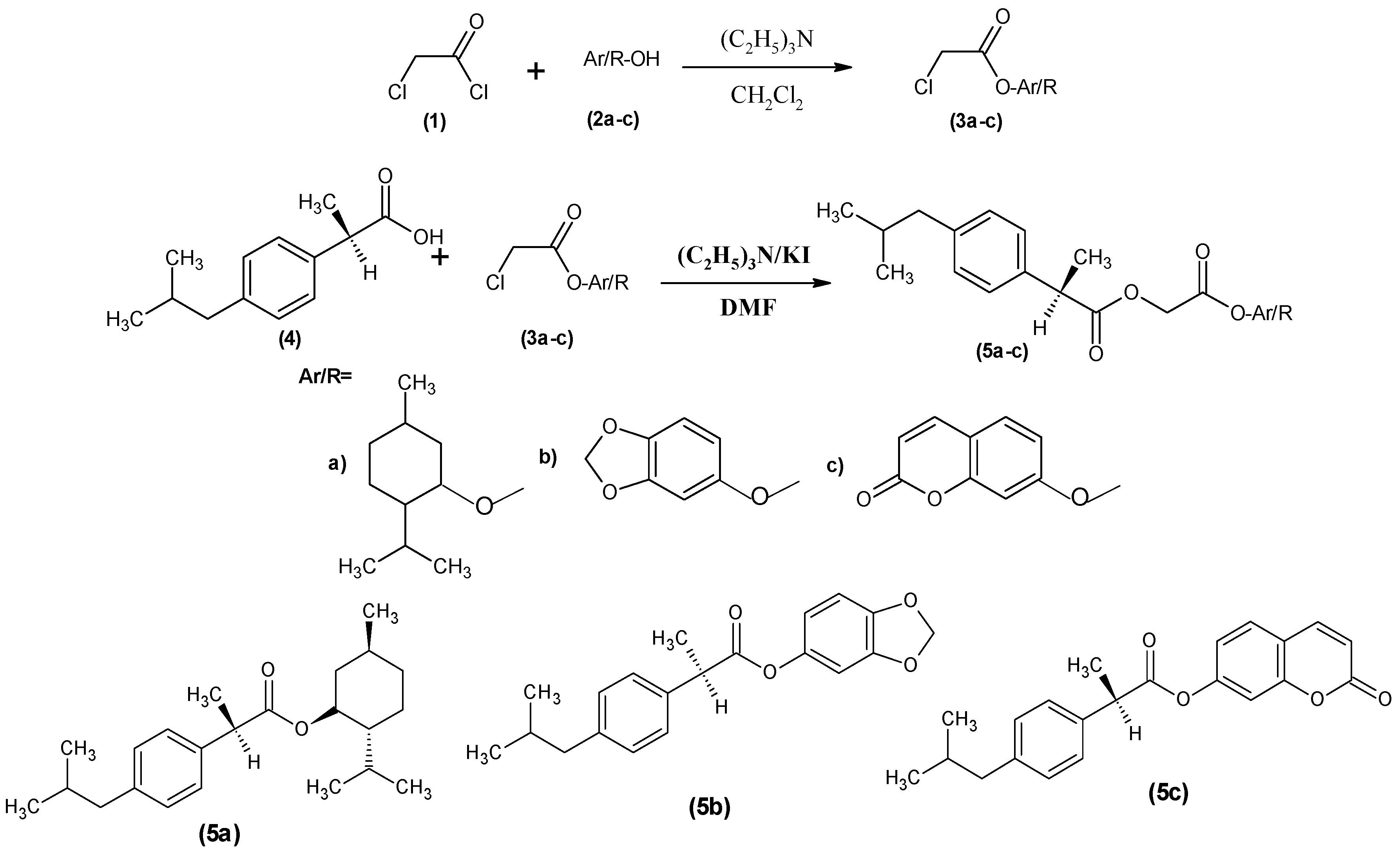
2.1. Synthesis of Prodrugs
2.2. Hydrolysis Studies of Mutual Prodrugs Dexibuprofen
2.3. Pharmacology
Anti-Inflammatory Activity
2.4. Analgesic Activity
2.5. Antipyretic Activity
2.6. Ulcerogenic Activity
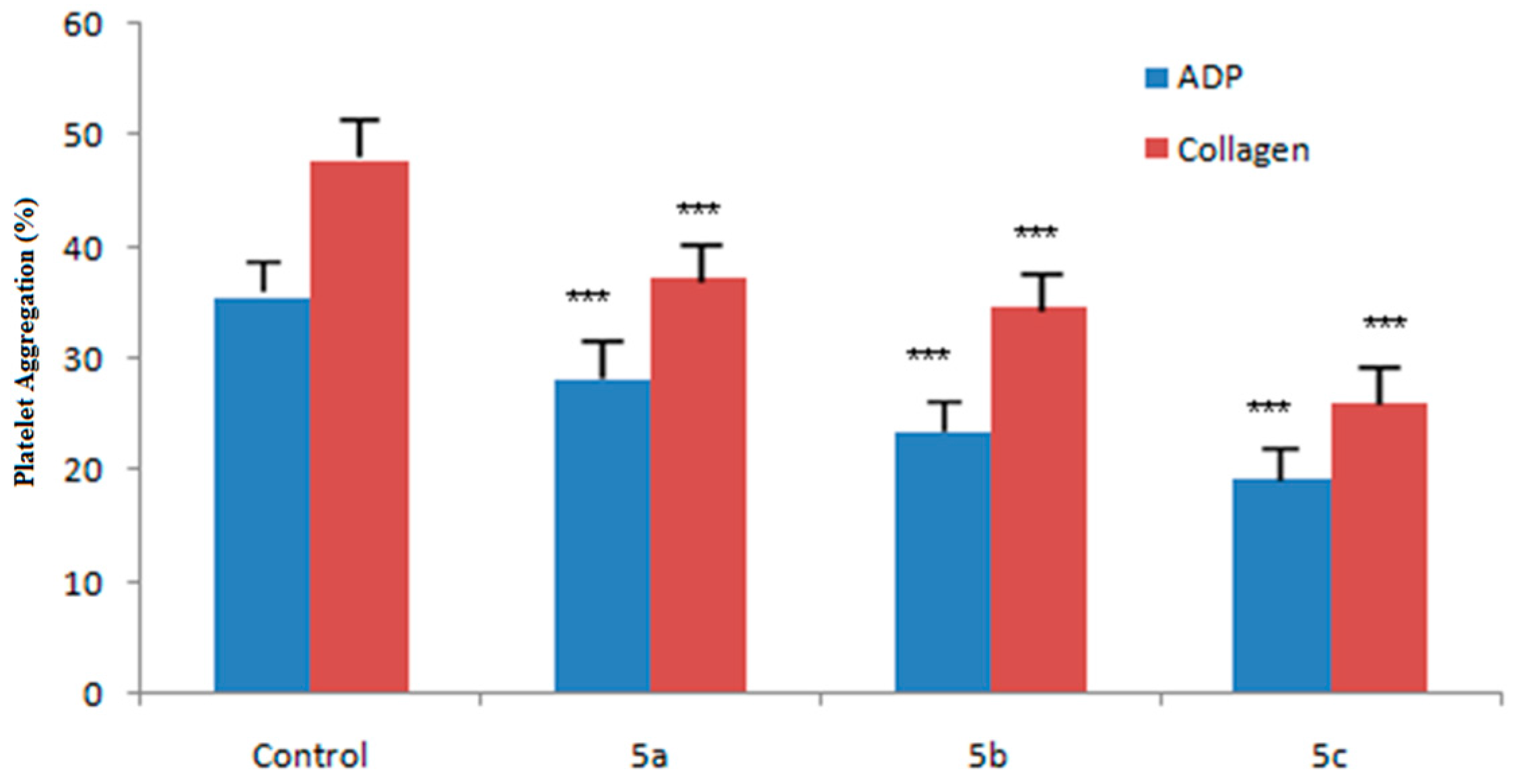
2.7. Ex Vivo Antiplatelet Aggregation Activity
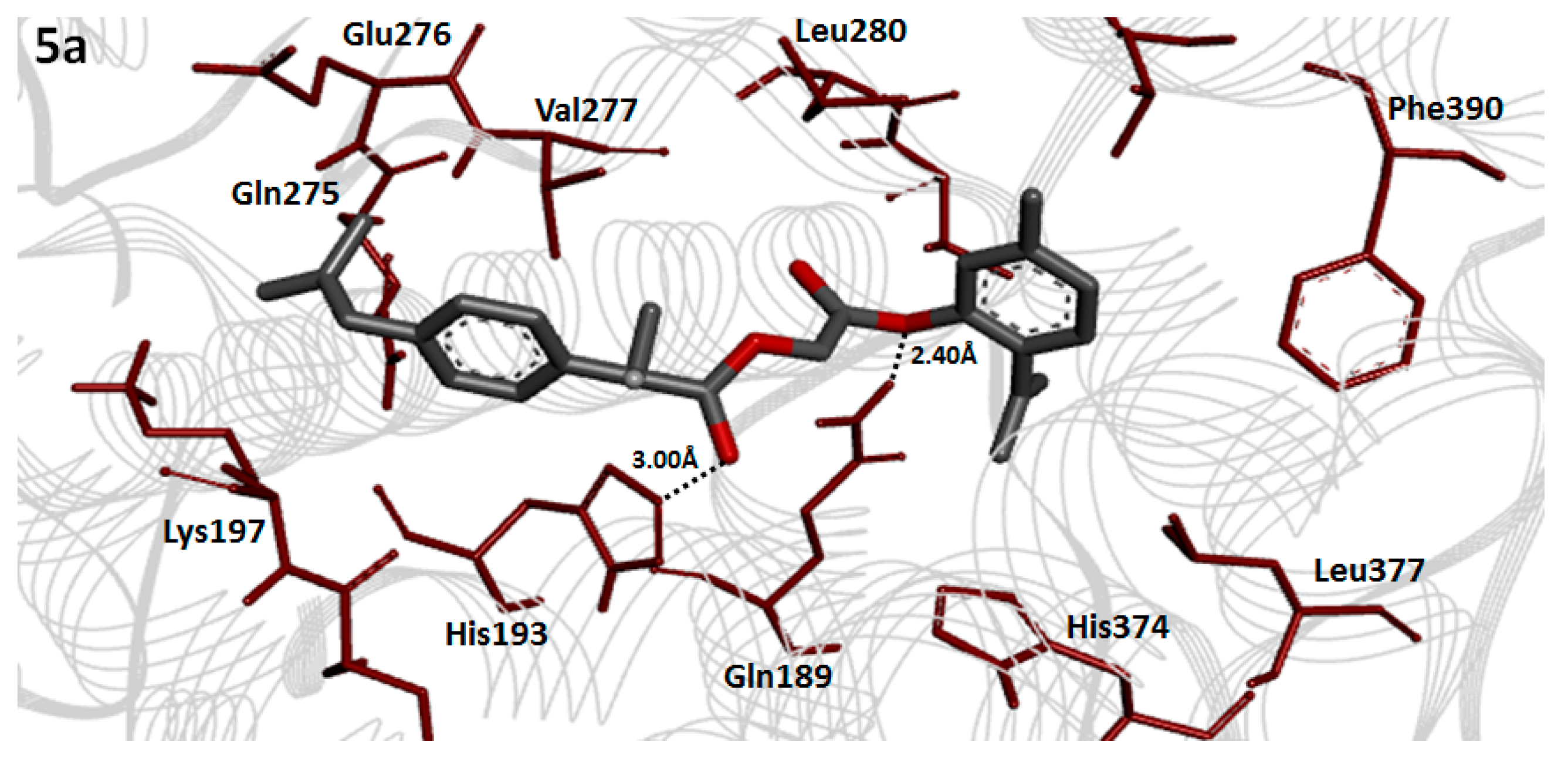
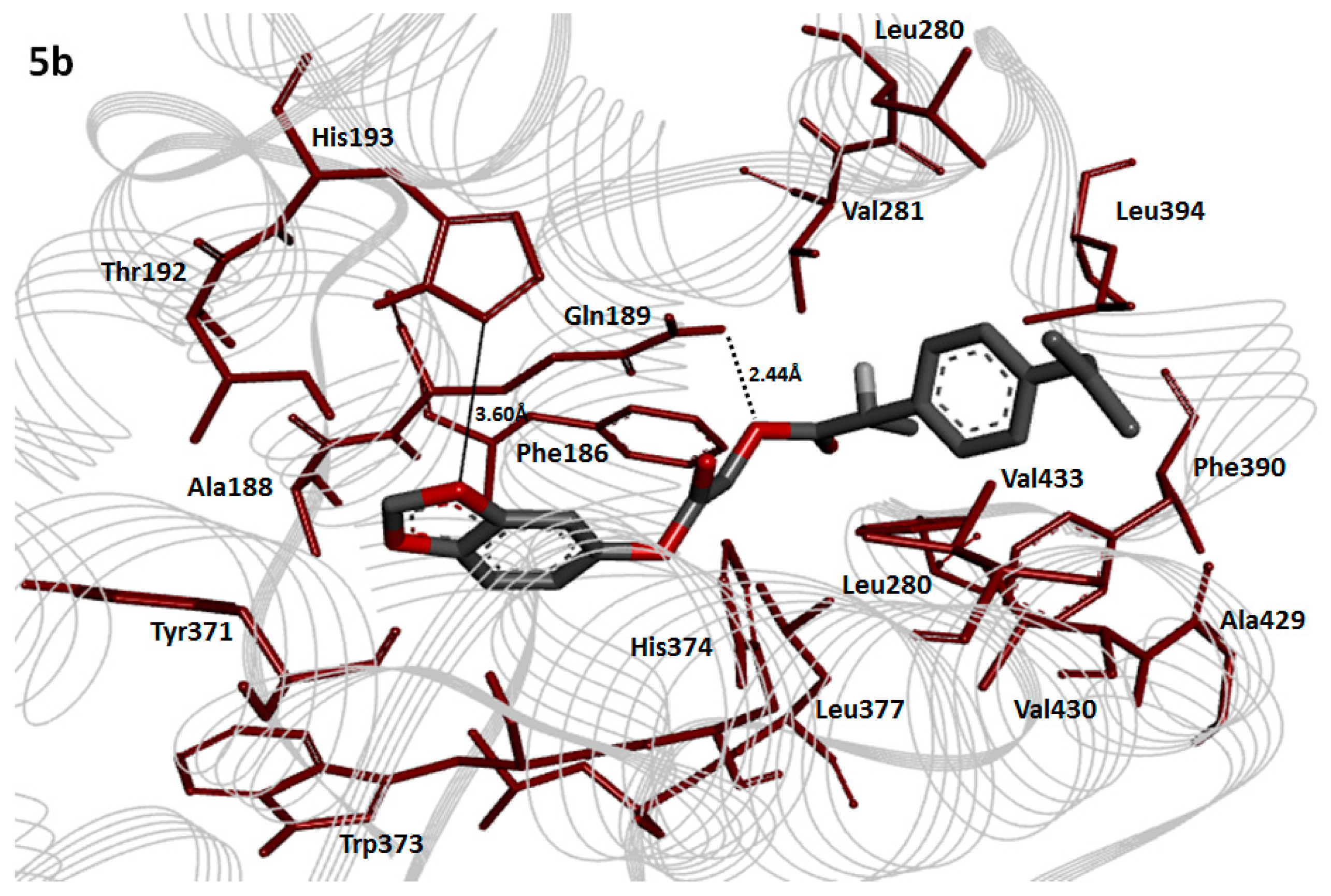
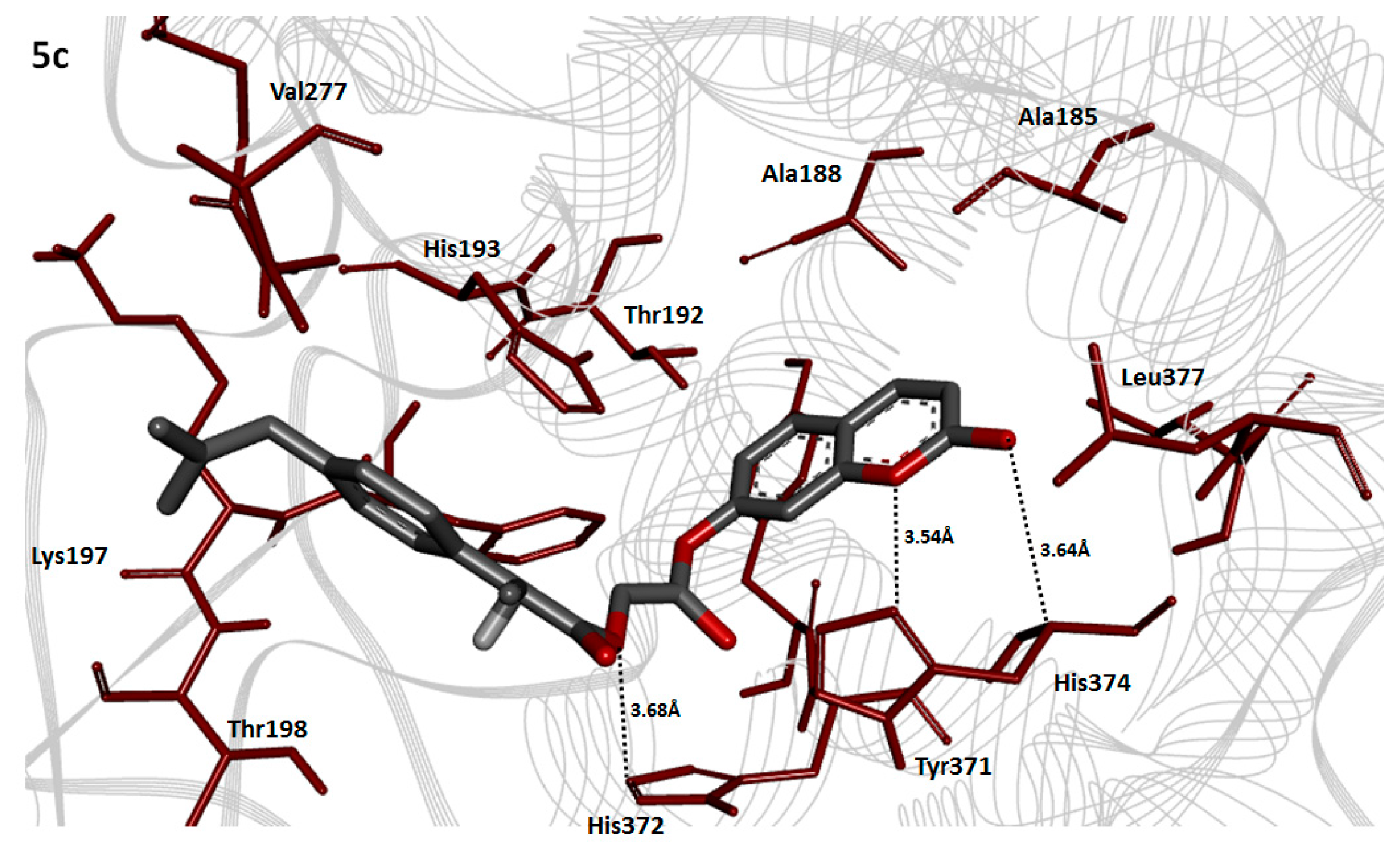
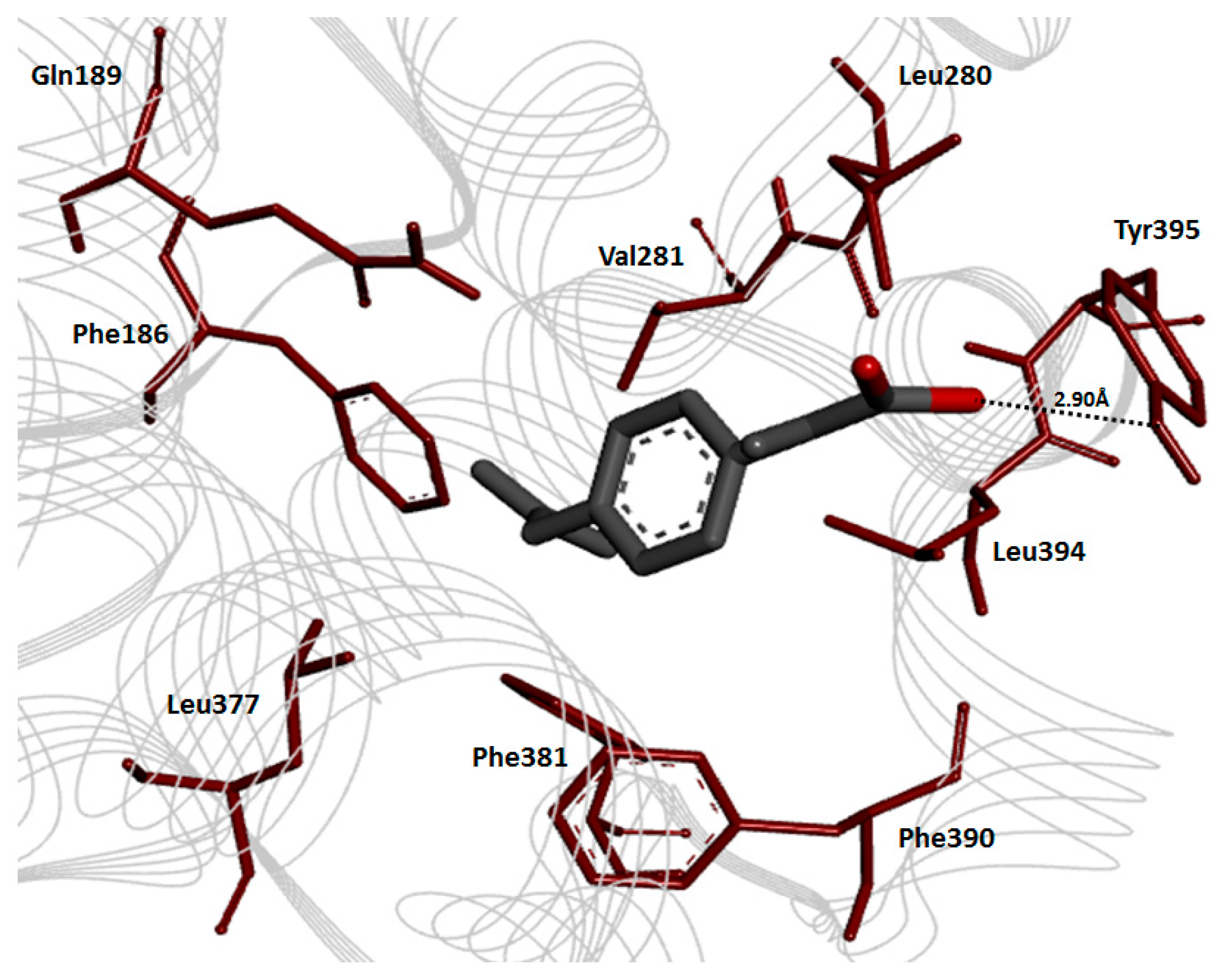
2.8. Molecular Docking Analysis
Cyclooxygenase-2 Evaluation
2.9. Prodrug Ligands Evaluation
2.10. Binding Energy Analysis
2.11. Hydrogen Bonding Analysis
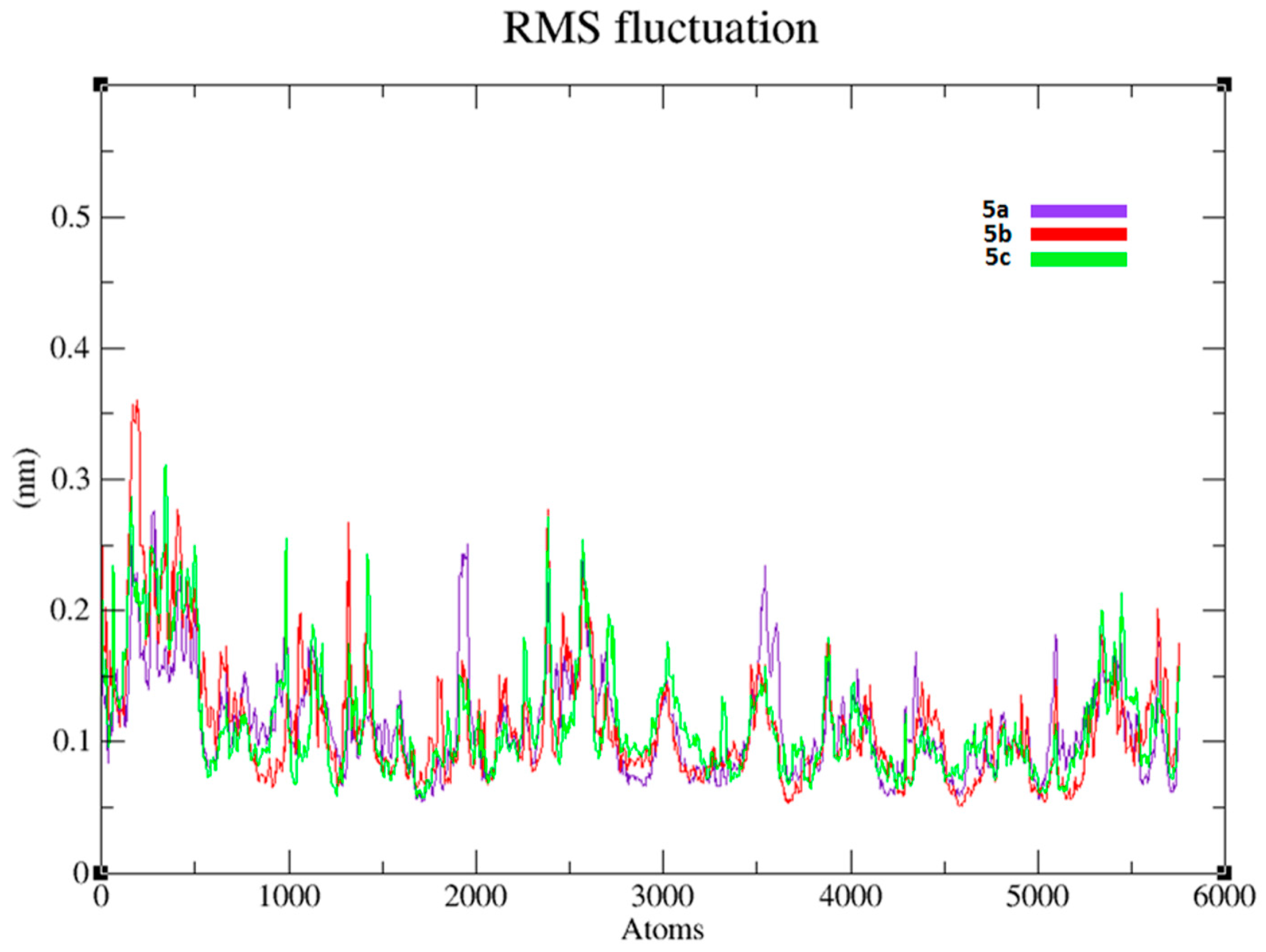
2.12. Molecular Dynamics (MD) Simulations Analysis
3. Material and Methods
3.1. Chemicals
3.2. Experimental
3.3. Synthesis of Antioxidant Chloroacetyl Derivatives (3a–c)
General Procedure
3.4. Synthesis of Dexibuprofen Antioxidant Mutual Prodrugs 5a–c
General Procedure
3.5. In Vitro Hydrolysis
3.6. Pharmacology
Animals
3.7. Animal Care
3.7.1. Temperature and Humidity
3.7.2. Ventilation and Air Quality
3.7.3. Drug Administration
3.8. Anti-Inflammatory Activity
Carrageenan Induced Paw Edema in Mice
3.9. Albumin Induced Inflammation in Mice
3.10. Analgesic Activity
3.10.1. Acetic Acid Induced Writhing in Mice
3.10.2. Formalin Induced Licking in Mice
3.11. Antipyretic Activity
3.12. Ulcerogenic Studies
3.13. Ex Vivo Antiplatelet Aggregation Activity
3.14. Statistical Analysis
3.15. Protein Structure from PDB
3.16. Prodrugs Preparation
3.17. Grid Generation and Molecular Docking
3.18. Molecular Dynamics (MD) Simulation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gabard, B.; Nirnberge, G.; Schiel, H.; Mascher, H.; Kikuta, C.; Mayer, J.M. Comparison of the bioavailability of dexibuprofen administered alone or as part of racemic ibuprofen. Eur. J. Clin. Pharmacol. 1995, 48, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Bonabello, A.; Galmozzi, M.R.; Canaparo, R.; Isaia, G.C.; Serpe, L.; Muntoni, E.; Zara, G.P. Dexibuprofen (S(+)-isomer ibuprofen) reduces gastric damage and improves analgesic and antiinflammatory effects in rodents. Anaesth. Analg. 2004, 97, 402–408. [Google Scholar] [CrossRef]
- Walser, S.; Hruby, R.; Hesse, E.; Heinzl, H.; Mascher, H. Preliminary toxicokinetic study with different crystal forms of S(+)-ibuprofen (dexibuprofen) and R,S-ibuprofen in rats. Arzneimittelforschung 1997, 47, 750–754. [Google Scholar] [PubMed]
- Kaehler, S.T.; Phleps, W.; Hesse, E. Dexibuprofen: Pharmacology, therapeutic uses and safety. Inflamm. Pharm. 2003, 11, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Benu, M.; Sharma, P.D. Design, synthesis and evaluation of diclofenac-antioxidant mutual prodrugs as safer NSAIDs. Indian J. Chem. 2009, 48B, 1279–1287. [Google Scholar]
- Polonia, J. Interaction of antihypertensive drugs with antiinflammatory drugs. J. Cardiol. 1997, 88, 47–51. [Google Scholar]
- Rasheed, A.; Aishwarya, K.; Basha, N.; Reddy, B.S.; Swetha, A. Dexibuprofen-dextran macromolecular prodrugs: Synthesis, characterization and pharmacological evaluation. Der Pharm. Chem. 2009, 2, 124–132. [Google Scholar]
- Bernard, S. Profile and mechanism of gastrointestinal and other side effects of non-steroidal antiinflammatory drugs (NSAIDs). Am. J. Med. 1999, 17, 27–35. [Google Scholar]
- Peng, Y.S.; Lin, S.C.; Huang, S.J.; Wang, Y.M.; Lin, Y.J.; Wang, L.F.; Chen, J.S. Chondroitin sulfate-based anti-inflammatory macromolecular Prodrugs. Eur. J. Pharm. Sci. 2006, 1, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Zgoda, M.M.; Lukosek, M.; Nachajski, M.J. Micellarsolubilization of selected non-steroidal therapeutic agents by new surfaceactive agents of the class of the products of oxyethylation of ursodeoxycholic acid. Polim. Med. 2006, 4, 13–30. [Google Scholar]
- Zhao, X.; Tao, X.; Wei, D.; Song, Q. Pharmacological activity and hydrolysis behavior of novel ibuprofen glucopyranoside conjugates. Eur. J. Med. Chem. 2006, 11, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Arun, R.; Ashok, K.C.K. Synthesis, hydrolysis and pharmacodynamic profiles of novel prodrugs of mefenamic acid. Int. J. Curr. Pharm. Res. 2009, 1, 47–55. [Google Scholar]
- Shanbhag, V.R.; Crider, A.M.; Gokhale, R.; Harpalani, A.; Dick, R.M. Ester and amide prodrugs of ibuprofen and naproxen: Synthesis, anti-inflammatory activity and gastrointestinal toxicity. J. Pharm. Sci. 2006, 81, 149–154. [Google Scholar] [CrossRef]
- Khan, M.S.Y.; Akhter, M. Synthesis, pharmacological activity and hydrolyticbehavior of glyceride prodrugs of ibuprofen. Eur. J. Med. Chem. 2005, 40, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.R.; Kulkarni, A.A.; Ghulekar, S.P. Synthesis, pharmacological activity and hydrolytic behavior of ethylenediamine and benzathine conjugates of ibuprofen. Eur. J. Med. Chem. 2008, 43, 2819–2823. [Google Scholar] [CrossRef] [PubMed]
- Shaaya, O.; Magora, A.; Sheskin, T.; Abraham, N.K.; Domb, J. Anhydride prodrugs for nonsteroidal anti-inflammatory drugs. Pharm. Res. 2003, 20, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Banekovich, C.; Ott, I.; Koch, T.; Matuszczak, B.; Gust, R. Synthesis and biological activities of novel dexibuprofen tetraacetyl riboflavin conjugate. Bioorg. Med. Chem. Lett. 2007, 17, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, Z.; Imran, M.; Amin, S. Synthesis, characterization and in vitro hydrolysis studies of ester and amide prodrugs of dexibuprofen. Med. Chem. Res. 2012, 21, 3361–3368. [Google Scholar] [CrossRef]
- Sharma, P.D.; Kaur, G.; Kansal, S.; Chandiran, S.K. Mutual prodrugs of 4-biphenylacetic acid and phytophenolics as safer NSAIDs: Synthetic and spectral studies. Indian J. Chem. Sect. B 2004, 43, 2159–2164. [Google Scholar]
- Ashraf, Z.; Rafiq, M.; Seo, S.Y.; Kwon, K.S.; Babar, M.M.; Zaidi, N.S.S. Kinetic and in silico studies of novel hydroxy-based thymol analogues as inhibitors of mushroom tyrosinase. Eur. J. Med. Chem. 2015, 98, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, Y.K.; Yasukawa, S.; Kitanaka, M.; Khan, T.; Evans, F.J. Effect of methanol extract from flower petals of Tagetes patella L. on acute and chronic inflammation model. Phytother. Res. 2002, 16, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R. Inhibition of Prostaglandin Synthesis as a Mechanism of Action for Aspirin-like Drugs. Nat. New Biol. 1971, 231, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Linardi, A.; Costa, S.K.P.; Desilva, G.R.; Antunes, E. Involvement of kinins, mast cells and sensory neurons in the plasma exudation and paw edema induced by staphylococcal entrotoxin B in the mouse. Eur. J. Pharmacol. 2002, 399, 235–242. [Google Scholar] [CrossRef]
- Vasudevan, M.; Gunman, K.K.; Parle, M. Antinociceptive and anti-inflammatory effects of the spesiapopulnea bark extract. J. Ethnopharmacol. 2007, 109, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Rang, H.P.; Dale, M.M.; Ritter, J.M.; Flower, R.J. Rang & Dale′s Pharmacology, 6th ed.; Landon: Bethesda, MD, USA, 2007. [Google Scholar]
- Bentley, G.A.; Newton, S.H.; Star, J.B. Studies on the anti-nociceptive action of α-agonist drugs and their interaction with opoid mechanism. Br. J. Pharmacol. 1983, 79, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Deraedt, R.; Jongney, S.; Delevalcee, F.; Falhout, M. Release of prostaglandin E and F in an analgesic reaction and its inhibition. Eur. J. Pharmacol. 1980, 51, 17–24. [Google Scholar] [CrossRef]
- Dhara, A.K.; Sube, V.; Sen, T.; Pal, A.N.; Chandhiri, A.K. Preliminary studies on the anti-inflammatory and analgesic activity of thmethanolic frictions of the root extract of Tragia involucrate. J. Ethnopharmacol. 2000, 72, 265–268. [Google Scholar] [CrossRef]
- Shibata, M.; Ohkubo, T.; Takahashi, H.; Inoki, R. Modified formalin test: Characteristic biphasic pain response. Pain 1989, 38, 347–352. [Google Scholar] [CrossRef]
- Ghannadi, A.; Hajhashem, V.; Jafarabadi, H. An investigation of the analgesic and anti-inflammatory effects of Nigella sativa seed polyphenols. J. Med. Food 2005, 8, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Rititid, W.; Ruangsang, P.; Reanmongkol, W.; Wongnawa, M. Studies of the anti-inflammatory and antipyretic activities of the methanolicextract of Piper sarmentosumRoxb.Leaves in rats. Songklanakarin J. Sci. Technol. 2007, 6, 1519–1526. [Google Scholar]
- Ayoub, S.S.; Colville, N.P.R.; Willoughby, D.A.; Botting, R.M. The involvement of a cyclooxygenase-1 gene-derived protein in the antinociceptive action of paracetamol in mice. Eur. J. Pharmacol. 2006, 538, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharan, D.V.; Roos, H.; Evanson, K.I.; Tomsik, N.K.; Elton, J.; Simmons, T. Cox-3, a cox-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure and expression. Proc. Natl. Acad. Sci. USA 2002, 139, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Kadam, R.U.; Roy, N. Recent trends in drug-likeness prediction: A comprehensive Review of in silicomethods. Indian J. Pharm. Sci. 2007, 69, 609–615. [Google Scholar]
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem. Neurosci. 2012, 3, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Bakht, M.A.; Yar, M.S.; Abdel-Hamid, S.G.; Al Qasoumi, S.I.; Samad, A. Molecular properties prediction, synthesis and antimicrobial activity of some newer oxadiazole derivatives. Eur. J. Med. Chem. 2010, 45, 5862–5869. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv. Drug Deliv. Rev. 2015, 86, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, P.B.; Yadav, A.R.; Gore, M.G. Concept of drug likeness in pharmaceutical research. Int. J. Pharm. Biol. Sci. 2015, 6, 142–154. [Google Scholar]
- Amaravani, M.; Prasad, N.K.; Ramakrishna, V. COX-2 structural analysis and docking studies with gallic acid structural analogues. SpringerPlus 2012, 1, 58. [Google Scholar] [CrossRef] [PubMed]
- Elengoe, A.; Naser, M.A.; Hamdan, S. Modeling and docking studies on novel mutants (K71L and T204V) of the ATPase domain of human heat shock 70kDa protein 1. Int. J. Mol. Sci. 2014, 15, 6797–6814. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [PubMed]
- Nwafor, P.A.; Okwuasaba, F.K. Antinociceptive and anti-inflammatory effects of methanolic extract of Asparagus pubescans roots in rodents. J. Ethanopharmacol. 2003, 84, 125–129. [Google Scholar] [CrossRef]
- Akah, P.A.; Nwambie, A.L. Evaluation of Nigerian traditional medicines: Plants used for rheumatic disorders. J. Ethanopharmacol. 1994, 42, 179–182. [Google Scholar] [CrossRef]
- Koster, R.; Anderson, M.; Debeer, J.M. Acetic acid used for analgesic screening. Fed. Proc. 1959, 18, 412–416. [Google Scholar]
- Correa, C.R.; Calixto, J.B. Evidence of participation of β1 and β2 Kinin receptors in formation-induced nociceptive response in mouse. Br. J. Pharmacol. 1993, 110, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Sakande, J.; Nacoulma, O.G.; Nikiema, J.B.; Lompo, M.; Bassene, E.; Guissou, I.P. Etude de1′effet antipyretique d′extraits des inflorescences malesduronier Borassusaethiopum Mart (Arecaceae). Med. Afr. N. 2004, 51, 280–282. [Google Scholar]
- Ezer, E.; Palosi, E.; Hajos, G.; Szporny, L. Antagonism of the gastrointestinal ulcerogenic effect of some nonsteroidal anti-inflammatory agents by sodium salicylate. J. Pharm. Pharmacol. 1976, 28, 655–656. [Google Scholar] [CrossRef] [PubMed]
- Vogel, W.H. Analgesic, anti-inflammatory, and antipyretic activity. In Drug Discovery and Evaluation; Springer: Berlin/Heidelberg, Germany, 1997; pp. 360–420. [Google Scholar]
- Majumdar, B.; Chaudhari, S.G.R.; Ray, A.; Bandyopadhyay, S.K. Effect of ethanol extract of Piper betle Linn leaf on healing of NSAID-induced experimental ulcer—a novel role of free radical scavenging action. Indian J. Exp. Biol. 2003, 41, 311–315. [Google Scholar] [PubMed]
- Kimura, Y.; Tani, T.; Kanbe, T.; Watanabe, K. Effect of cilostazol on platelet aggregation and experimental thrombosis. Drug Res. 1985, 35, 1144–1149. [Google Scholar]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., 3rd; de Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: Phi,psi and Cβ deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [PubMed]
- Studio. Discovery, version 2.1; Accelrys: San Diego, CA, USA, 2008.
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; van Gunsteren, W.F. Abiomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Schüttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dommert, F.; Holm, C. Optimizing working parameters of the smooth particle mesh Ewald algorithm in terms of accuracy and efficiency. J. Chem. Phys. 2010, 133, 034117. [Google Scholar] [CrossRef] [PubMed]
- Amiri, S.; Sansom, M.S.; Biggin, P.C. Molecular dynamics studies of AChBP with nicotine and carbamylcholine: The role of water in the binding pocket. Protein Eng. Des. Sel. 2007, 20, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Labik, S.; Smith, W.R. Scaled Particle Theory and the Efficient Calculation of the Chemical-Potential of Hard-Spheres in the Nvt Ensemble. Mol. Simul. 1994, 12, 23–31. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serial. No. | Inflammation at 0 h (mm) | Inflammation at 1 h (mm) | Inflammation at 2 h (mm) | Inflammation at 3 h (mm) | Inflammation at 4 h (mm) |
|---|---|---|---|---|---|
| Dose | Mean ± SEM | Mean ± SEM | Mean ± SEM | Mean ± SEM | Mean ± SEM |
| Control 10 mg/kg | 4.17 ± 0.08 | 4.40 ± 0.10 | 4.6 ± 0.06 | 4.85 ± 0.06 | 5.07 ± 0.06 |
| Dexibuprofen 20 mg/kg | 4.37 ± 0.06 | 4.35 ± 0.06 ns (1.1%) | 4.07 ± 0.02 *** (11.71%) | 3.90 ± 0.04 *** (19.58%) | 3.75 ± 0.06 *** (26.03%) |
| 5a 20 mg/kg | 4.42 ± 0.05 | 4.50 ± 0.04 ns (−2.27%) | 4.25 ± 0.06 ** (7.80%) | 4.12 ± 0.06 *** (15.05%) | 3.92 ± 0.02 *** (22.68%) |
| 5b 20 mg/kg | 4.37 ± 0.04 | 4.22 ± 0.1 ns (4.09%) | 4.25 ± 0.06 ** (7.80%) | 4.15 ± 0.02 *** (14.43%) | 3.92 ± 0.05 *** (22.68%) |
| 5c 20 mg/kg | 4.32 ± 0.04 | 4.22 ± 0.04 ns (4.09%) | 4.05 ± 0.02 *** (12.14%) | 3.80 ± 0.04 *** (21.64%) | 3.60 ± 0.04 *** (28.99%) |
| Treatment/Dose | Inflammation at 0 h (mm) | Inflammation at 1 h (mm) | Inflammation at 2 h (mm) | Inflammation at 3 h (mm) |
|---|---|---|---|---|
| Dose | Mean ± SEM | Mean ± SEM | Mean ± SEM | Mean ± SEM |
| Control 10 mL/kg | 4.35 ± 0.05 | 4.40 ± 0.10 | 4.80 ± 0.10 | 5.22 ± 0.07 |
| Dexibuprofen 20 mg/kg | 4.05 ± 0.41 | 3.20 ± 0.04 *** (27.27%) | 3.42 ± 0.20 *** (28.75%) | 3.00 ± 0.00 *** (42.52%) |
| 5a 20 mg/kg | 4.67 ± 0.08 | 3.70 ± 0.16 * (15.90%) | 3.37 ± 0.20 *** (29.79%) | 3.30 ± 0.04 *** (36.78%) |
| 5b 20 mg/kg | 4.15 ± 0.32 | 3.95 ± 0.02 ns (10.22%) | 3.40 ± 0.20 *** (29.16%) | 3.17 ± 0.11 *** (39.27%) |
| 5c 20 mg/kg | 4.60 ± 0.33 | 3.12 ± 0.06 *** (29.09%) | 3.02 ± 0.06 *** (37.08%) | 2.92 ± 0.07 *** (44.06%) |
| Treatment/Dose | No. of Writhing | Lickings Time |
|---|---|---|
| Dose | Mean ± SEM | Mean ± SEM |
| Control 10 mL/kg | 37.20 ± 1.10 | 2.83 ± 0.26 |
| Dexibuprofen 20 mg/kg | 17.06 ± 1.08 *** (54.12%) | 1.60 ± 0.22 *** (43.46%) |
| 5a 20 mg/kg | 15.00 ± 1.29 *** (59.67%) | 0.70 ± 0.22 *** (75.26%) |
| 5b 20 mg/kg | 17.50 ± 0.64 *** (52.95%) | 0.98 ± 0.17 *** (65.37%) |
| 5c 20 mg/kg | 18.25 ± 0.85 *** (50.94%) | 1.31 ± 0.07 *** (53.71%) |
| Treatment/Dose | Temperature before Induction (°F) | After 24 h of Induction (°F) | 1 h (°F) | 2 h (°F) | 3 h (°F) |
|---|---|---|---|---|---|
| Dose | Mean ± SEM | Mean ± SEM | Mean ± SEM | Mean ± SEM | Mean ± SEM |
| Control (10 mL/kg) | 95.17 ± 0.34 | 96.02 ± 0.44 | 96.25 ± 0.39 | 96.22 ± 0.39 | 96.22 ± 0.39 |
| Dexibuprofen (20 mg/kg) | 95.20 ± 0.21 | 95.77 ± 0.26 | 95.25 ± 0.37 ns | 95.02 ± 0.35 * | 94.97 ± 0.31 * |
| 5a (20 mg/kg) | 94.37 ± 0.04 | 95.14 ± 0.02 | 94.97 ± 0.06 * | 94.62 ± 0.08 ** | 94.37 ± 0.08 *** |
| 5b (20 mg/kg) | 94.25 ± 0.10 | 95.47 ± 0.19 | 94.87 ± 0.06 ** | 94.5 ± 0.12 *** | 94.17 ± 0.06 *** |
| 5c (20 mg/kg) | 98.95 ± 0.04 | 102.1 ± 0.96 | 98.35 ± 0.05 *** | 97.65 ± 0.15 ** | 97.25 ± 0.08 ns |
| Serial. No. | Compounds | Ulcer Index (Mean±SEM) |
|---|---|---|
| Control group | CMC * | 0.37 ± 0.37 |
| 1 | Dexibuprofen | 2.89 ± 0.63 |
| 2 | 5a | 1.55 ± 0.09 ** |
| 3 | 5b | 1.34 ± 0.06 ** |
| 4 | 5c | 1.61 ± 0.58 * |
| Prodrugs | Docking Energy (kcal/mol) | Binding Pocket Residues |
|---|---|---|
| 5a | −8.90 | Gln189, His193, Lys197, Leu377, Phe390, Val281, Leu280, val273, Glu276, Gln275, His |
| 5b | −9.90 | Gln189, His193, Thr192, Ala188, Phe186, Tyr371, Trp373, His374, Leu276, Leu377, Ala429, Val430,Val433, Phe390, Leu394, Val281, Leu280 |
| 5c | −9.40 | His193, Thr192, Ala188, Phe185, Val277, Lys197, Thr198, His372, Tyr371, His374, Leu377, Ala185 |
| Dexibuprofen | −8.60 | Gln189, Phe390, Val281, Leu263, Leu394 and Leu377 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashraf, Z.; Alamgeer; Rasool, R.; Hassan, M.; Ahsan, H.; Afzal, S.; Afzal, K.; Cho, H.; Kim, S.J. Synthesis, Bioevaluation and Molecular Dynamic Simulation Studies of Dexibuprofen–Antioxidant Mutual Prodrugs. Int. J. Mol. Sci. 2016, 17, 2151. https://doi.org/10.3390/ijms17122151
Ashraf Z, Alamgeer, Rasool R, Hassan M, Ahsan H, Afzal S, Afzal K, Cho H, Kim SJ. Synthesis, Bioevaluation and Molecular Dynamic Simulation Studies of Dexibuprofen–Antioxidant Mutual Prodrugs. International Journal of Molecular Sciences. 2016; 17(12):2151. https://doi.org/10.3390/ijms17122151
Chicago/Turabian StyleAshraf, Zaman, Alamgeer, Raqiqatur Rasool, Mubashir Hassan, Haseeb Ahsan, Samina Afzal, Khurram Afzal, Hongsik Cho, and Song Ja Kim. 2016. "Synthesis, Bioevaluation and Molecular Dynamic Simulation Studies of Dexibuprofen–Antioxidant Mutual Prodrugs" International Journal of Molecular Sciences 17, no. 12: 2151. https://doi.org/10.3390/ijms17122151
APA StyleAshraf, Z., Alamgeer, Rasool, R., Hassan, M., Ahsan, H., Afzal, S., Afzal, K., Cho, H., & Kim, S. J. (2016). Synthesis, Bioevaluation and Molecular Dynamic Simulation Studies of Dexibuprofen–Antioxidant Mutual Prodrugs. International Journal of Molecular Sciences, 17(12), 2151. https://doi.org/10.3390/ijms17122151