
NAFLD and NASH in HCV Infection: Prevalence and Significance in Hepatic and Extrahepatic Manifestations

Abstract
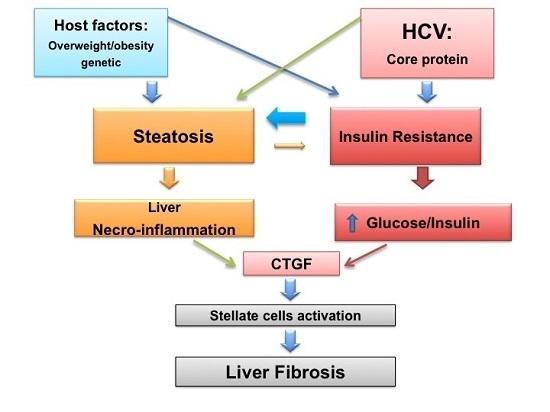
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Prevalence of NAFLD/NASH and Associated Conditions in Chronic HCV Infection
3. HCV-Induces Steatosis: Is It a Finalistic Condition?
4. HCV-Associated Steatosis and Progression of Liver Damage
5. HCV-Associated Steatosis, Diabetes, Metabolic Syndrome, and Atherosclerosis
6. HCV-Associated Steatosis and Response to Antiviral Treatments
7. Conclusions
Author Contributions
Conflicts of Interest
References
- Christopher, D.; Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, L.E.; Gambardella, M.; Andreana, A.; Tripodi, M.F.; Utili, R.; Ruggiero, G. Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology 2001, 33, 1358–1364. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, L.E.; Utili, R.; Ruggiero, G. Body composition and hepatic steatosis as precursors of fibrosis in chronic hepatitis C patients. Hepatology 1999, 30, 1530–1531. [Google Scholar] [CrossRef] [PubMed]
- Rubbia-Brandt, L.; Quadri, R.; Abid, K.; Giostra, E.; Malé, P.J.; Mentha, G.; Spahr, L.; Zarski, J.P.; Borisch, B.; Hadengue, A.; et al. Hepatocyte steatosis is a cytopathic effect of hepatitis C virus genotype 3. J. Hepatol. 2000, 33, 106–115. [Google Scholar] [CrossRef]
- Poynard, T.; Ratziu, V.; McHutchison, J.; Manns, M.; Goodman, Z.; Zeuzem, S.; Younossi, Z.; Albrecht, J. Effect of treatment with peginterferon or interferon alfa-2b and ribavirin on steatosis in patients infected with hepatitis C. Hepatology 2003, 38, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Patton, H.M.; Patel, K.; Behling, C.; Bylund, D.; Blatt, L.M.; Vallée, M.; Heaton, S.; Conrad, A.; Pockros, P.J.; McHutchison, J.G. The impact of steatosis on dise. J. Hepatol. 2004, 40, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Hickman, I.J.; Powell, E.E.; Prins, J.B.; Clouston, A.D.; Ash, S.; Purdie, D.M.; Jonsson, J.R. In overweight patients with chronic hepatitis C, circulating insulin is associated with hepatic fibrosis: Implications for therapy. J. Hepatol. 2003, 39, 1042–1048. [Google Scholar] [CrossRef]
- Zubair, A.; Jamal, S.; Mubarik, A.; Saudi, J. Morphometric analysis of hepatic steatosis in chronic hepatitis C infection. Saudi J. Gastroenterol. 2009, 15, 11–14. [Google Scholar] [PubMed]
- Pazienza, V.; Clément, S.; Pugnale, P.; Conzelman, S.; Foti, M.; Mangia, A.; Negro, F. The hepatitis C virus core protein of genotypes 3A and 1b downregulates insulin receptor substrate 1 through genotype-specific mechanisms. Hepatology 2007, 45, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.J.; Luo, J.C.; Chu, C.W.; Lai, C.R.; Lu, C.L.; Tsay, S.H.; Wu, J.C.; Chang, F.Y.; Lee, S.D. Hepatic steatosis in chronic hepatitis C virus infection: Prevalence and clinical correlation. J. Gastroenterol. Hepatol. 2001, 16, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Zeuzem, S.; Hultcrantz, R.; Bourliere, M.; Goeser, T.; Marcellin, P.; Sanchez-Tapias, J.; Sarrazin, C.; Harvey, J.; Brass, C.; Albrecht, J. Peginterferon alfa-2b plus ribavirin for treatment of chronic hepatitis C in previously untreated patients infected with HCV genotypes 2 or 3. J. Hepatol. 2004, 40, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Brunt, E.M.; Qazi, R.A.; Oliver, D.A.; Neuschwander-Tetri, B.A.; Di Bisceglie, A.M.; Bacon, B.R. Effect of significant histologic steatosis or steatohepatitis on response to antiviral therapy in patients with chronic hepatitis C. Clin. Gastroenterol. Hepatol. 2005, 3, 604–609. [Google Scholar] [CrossRef]
- Romero-Gómez, M.; del Mar Viloria, M.; Andrade, R.J.; Salmerón, J.; Diago, M.; Fernández-Rodríguez, C.M.; Corpas, R.; Cruz, M.; Grande, L.; Vázquez, L.; et al. Insulin resistance impairs sustained response rate to peginterferon plus ribavirin in chronic hepatitis C patients. J. Gastroenterol. 2005, 128, 636–641. [Google Scholar] [CrossRef]
- Kumar, D.; Farrell, G.C.; Fung, C.; George, J. Hepatitis C virus genotype 3 is cytopathic to hepatocytes: Reversal of hepatic steatosis after sustained therapeutic response. Hepatology 2002, 36, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Fujie, H.; Miyoshi, H.; Tsutsumi, T.; Tsukamoto, K.; Kimura, S.; Moriya, K.; Koike, K. Hepatitis C virus infection and diabetes: Direct involvement of the virus in the development of insulin resistance. Gastroenterology 2004, 126, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Adinolfi, L.E.; Loria, P.; Carulli, N.; Ruggiero, G.; Day, C.P. Steatosis and hepatitis C virus: Mechanisms and significance for hepatic and extrahepatic disease. Gastroenterology 2004, 126, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, L.E.; Zampino, R.; Restivo, L.; Lonardo, A.; Guerrera, B.; Marrone, A.; Nascimbeni, F.; Florio, A.; Loria, P. Chronic hepatitis C virus infection and atherosclerosis: Clinical impact and mechanisms. World J. Gastroenterol. 2014, 20, 3410–3417. [Google Scholar] [CrossRef] [PubMed]
- Vidali, M.; Tripodi, M.F.; Ivaldi, A.; Zampino, R.; Occhino, G.; Restivo, L.; Sutti, S.; Marrone, A.; Ruggiero, G.; Albano, E.; et al. Interplay between oxidative stress and hepatic steatosis in the progression of chronic hepatitis C. J. Hepatol. 2008, 48, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Adinolfi, L.E.; Restivo, L.; Ballestri, S.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Loria, P. Pathogenesis and significance of hepatitis C virus steatosis: An update on survival strategy of a successful pathogen. World J. Gastroenterol. 2014, 23, 7089–7103. [Google Scholar] [CrossRef] [PubMed]
- Mihm, S.; Fayyazi, A.; Hartmann, H.; Ramadori, G. Analysis of histopathological manifestations of chronic hepatitis C virus infection with respect to virus genotype. Hepatology 1997, 25, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J.; Carpenter, H.A.; Santrach, P.J.; Moore, S.B. Host- and disease-specific factors affecting steatosis in chronic hepatitis C. J. Hepatol. 1998, 29, 198–206. [Google Scholar] [CrossRef]
- Hourigan, L.F.; Macdonald, G.A.; Purdie, D.; Whitehall, V.H.; Shorthouse, C.; Clouston, A.; Powell, E.E. Fibrosis in chronic hepatitis C correlates significantly with body mass index and steatosis. Hepatology 1999, 29, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Serfaty, L.; Andreani, T.; Giral, P.; Carbonell, N.; Chazouillères, O.; Poupon, R. Hepatitis C virus induced hypobetalipoproteinemia: A possible mechanism for steatosis in chronic hepatitis C. J. Hepatol. 2001, 34, 428–434. [Google Scholar] [CrossRef]
- Monto, A.; Alonzo, J.; Watson, J.J.; Grunfeld, C.; Wright, T.L. Steatosis in chronic hepatitis C: Relative contributions of obesity, diabetes mellitus, and alcohol. Hepatology 2002, 36, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Westin, J.; Nordlinder, H.; Lagging, M.; Norkrans, G.; Wejstål, R. Steatosis accelerates fibrosis development over time in hepatitis C virus genotype 3 infected patients. J. Hepatol. 2002, 37, 837–842. [Google Scholar] [CrossRef]
- Hui, J.M.; Kench, J.; Farrell, G.C.; Lin, R.; Samarasinghe, D.; Liddle, C.; Byth, K.; George, J. Genotype-specific mechanisms for hepatic steatosis in chronic hepatitis C infection. J. Gastroenterol. Hepatol. 2002, 17, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Castéra, L.; Hézode, C.; Roudot-Thoraval, F.; Bastie, A.; Zafrani, E.S.; Pawlotsky, J.M.; Dhumeaux, D. Worsening of steatosis is an independent factor of fibrosis progression in untreated patients with chronic hepatitis C and paired liver biopsies. Gut 2003, 52, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Asselah, T.; Boyer, N.; Guimont, M.C.; Cazals-Hatem, D.; Tubach, F.; Nahon, K.; Daïkha, H.; Vidaud, D.; Martinot, M.; Vidaud, M.; et al. Liver fibrosis is not associated with steatosis but with necroinflammation in French patients with chronic hepatitis C. Gut 2003, 52, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Rubbia-Brandt, L.; Fabris, P.; Paganin, S.; Leandro, G.; Male, P.J.; Giostra, E.; Carlotto, A.; Bozzola, L.; Smedile, A.; Negro, F. Steatosis affects chronic hepatitis C progression in a genotype specific way. Gut 2004, 53, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.M.; Brancati, F.L.; Diehl, A.M. Nonalcoholic fatty liver disease. Gastroenterology 2002, 122, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.P.; Younossi, Z.M.; Speer, C.; Olano, A.; Gramlich, T.; Boparai, N. Chronic hepatitis C and superimposed nonalcoholic fatty liver disease. Liver 2001, 21, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Ramrakhiani, S.; Cordes, B.G.; Neuschwander-Tetri, B.A.; Janney, C.G.; Bacon, B.R.; di Bisceglie, A.M. Concurrence of histologic features of steatohepatitis with other forms of chronic liver disease. Mod. Pathol. 2003, 16, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Jeng, Y.M.; Chen, P.J.; Lai, M.Y.; Yang, H.C.; Huang, W.L.; Kao, J.H.; Chen, D.S. Influence of metabolic syndrome, viral genotype and antiviral therapy on superimposed fatty liver disease in chronic hepatitis C. Antivir. Ther. 2005, 10, 405–415. [Google Scholar] [PubMed]
- Bedossa, P.; Moucari, R.; Chelbi, E.; Asselah, T.; Paradis, V.; Vidaud, M.; Cazals-Hatem, D.; Boyer, N.; Valla, D.; Marcellin, P. Evidence for a role of nonalcoholic steatohepatitis in hepatitis C: A prospective study. Hepatology 2007, 46, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Solis-Herruzo, J.A.; Pérez-Carreras, M.; Rivas, E.; Fernández-Vázquez, I.; Garfia, C.; Bernardos, E.; Castellano, G.; Colina, F. Factors associated with the presence of nonalcoholic steatohepatitis in patients with chronic hepatitis C. Am. J. Gastroenterol. 2005, 100, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, J.R.; Barrie, H.D.; O’Rourke, P.; Clouston, A.D.; Powell, E.E. Obesity and steatosis influence serum and hepatic inflammatory markers in chronic hepatitis C. Hepatology 2008, 48, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Ballestri, S.; Adinolfi, L.E.; Violi, E.; Carulli, L.; Lombardini, S.; Scaglioni, F.; Ricchi, M.; Ruggiero, G.; Loria, P. Hepatitis C virus-infected patients are “spared” from the metabolic syndrome but not from insulin resistance. A comparative study of nonalcoholic fatty liver disease and hepatitis C virus-related steatosis. Can. J. Gastroenterol. 2009, 23, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Zampino, R.; Ingrosso, D.; Durante-Mangoni, E.; Capasso, R.; Tripodi, M.F.; Restivo, L.; Zappia, V.; Ruggiero, G.; Adinolfi, L.E. Microsomal triglyceride transfer protein (MTP)-493G/T gene polymorphism contributes to fat liver accumulation in HCV genotype 3 infected patients. J. Viral. Hepat. 2008, 10, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, L.E.; Ingrosso, D.; Cesaro, G.; Cimmino, A.; D’Antò, M.; Capasso, R.; Zappia, V.; Ruggiero, G. Hyperhomocysteinemia and the MTHFR C677T polymorphism promote steatosis and fibrosis in chronic hepatitis C patients. Hepatology 2005, 41, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Fargion, S. Patatin-like phospholipase domain containing-3 Ile148Met and fibrosis progression after liver transplantation. Hepatology 2011, 54, 1484. [Google Scholar] [CrossRef] [PubMed]
- Zampino, R.; Coppola, N.; Cirillo, G.; Boemio, A.; Grandone, A.; Stanzione, M.; Capoluongo, N.; Marrone, A.; Macera, M.; Sagnelli, E.; et al. Patatin-like phospholipase domain-containing 3 I148M variant is associated with liver steatosis and fat distribution in chronic hepatitis B. Dig. Dis. Sci. 2015, 60, 3005–3010. [Google Scholar] [CrossRef] [PubMed]
- Coppola, N.; Rosa, Z.; Cirillo, G.; Stanzione, M.; Macera, M.; Boemio, A.; Grandone, A.; Pisaturo, M.; Marrone, A.; Adinolfi, L.E.; et al. TM6SF2 E167K variant is associated with severe steatosis in chronic hepatitis C, regardless of PNPLA3 polymorphism. Liver Int. 2015, 35, 1959–1963. [Google Scholar] [CrossRef] [PubMed]
- Oem, J.K.; Jackel-Cram, C.; Li, Y.P.; Zhou, Y.; Zhong, J.; Shimano, H.; Babiuk, L.A.; Liu, Q. Activation of sterol regulatory element-binding protein 1C and fatty acid synthase transcription by hepatitis C virus non-structural protein 2. J. Gen. Virol. 2008, 89, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Perlemuter, G.; Sabile, A.; Letteron, P.; Vona, G.; Topilco, A.; Chrétien, Y.; Koike, K.; Pessayre, D.; Chapman, J.; Barba, G.; et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: A model of viral-related steatosis. FASEB J. 2002, 16, 185–194. [Google Scholar] [CrossRef] [PubMed]
- De Gottardi, A.; Pazienza, V.; Pugnale, P.; Bruttin, F.; Rubbia-Brandt, L.; Juge-Aubry, C.E.; Meier, C.A.; Hadengue, A.; Negro, F. Peroxisome proliferator-activated receptor-α and -γ mRNA levels are reduced in chronic hepatitis C with steatosis and genotype 3 infection. Aliment. Pharmacol. Ther. 2006, 23, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Burlone, M.E.; Budkowska, A. Hepatitis C virus cell entry: Role of lipoproteins and cellular receptors. J. Gen. Virol. 2009, 90, 1055–1070. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, S.B.; Chisari, F.V. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. USA 2005, 102, 2561–2566. [Google Scholar] [CrossRef] [PubMed]
- Westin, J.; Lagging, M.; Dhillon, A.P.; Norkrans, G.; Romero, A.I.; Pawlotsky, J.M.; Zeuzem, S.; Schalm, S.W.; Verheij-Hart, E.; Negro, F.; et al. Impact of hepatic steatosis on viral kinetics and treatment outcome during antiviral treatment of chronic HCV infection. J. Viral Hepat. 2007, 14, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, L.E.; Restivo, L.; Guerrera, B.; Sellitto, A.; Ciervo, A.; Iuliano, N.; Rinaldi, L.; Santoro, A.; li Vigni, G.; Marrone, A. Chronic HCV infection is a risk factor of ischemic stroke. Atherosclerosis 2013, 23, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Cross, T.J.; Quaglia, A.; Hughes, S.; Joshi, D.; Harrison, P.M. The impact of hepatic steatosis on the natural history of chronic hepatitis C infection. J. Viral Hepat. 2009, 16, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, U.; Arkkila, P.E.; Kärkkäinen, P.; Färkkilä, M.A. Effect of steatosis and inflammation on liver fibrosis in chronic hepatitis C. Liver Int. 2009, 29, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Leandro, G.; Mangia, A.; Hui, J.; Fabris, P.; Rubbia-Brandt, L.; Colloredo, G.; Adinolfi, L.E.; Asselah, T.; Jonsson, J.R.; Smedile, A.; et al. Relationship between steatosis, inflammation, and fibrosis in chronic hepatitis C: A meta-analysis of individual patient data. Gastroenterology 2006, 130, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Sterling, R.K.; Wegelin, J.A.; Smith, P.G.; Stravitz, R.T.; Luketic, V.A.; Fuchs, M.; Puri, P.; Shiffman, M.L.; Contos, M.A.; Mills, A.S.; et al. Similar progression of fibrosis between HIV/HCV-infected and HCV-infected patients: Analysis of paired liver biopsy samples. Clin. Gastroenterol. Hepatol. 2010, 8, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Matos, C.A.; Perez, R.M.; Pacheco, M.S.; Figueiredo-Mendes, C.G.; Lopes-Neto, E.; Oliveira, E.B.; Lanzoni, V.P.; Silva, A.E.; Ferraz, M.L. Steatosis in chronic hepatitis C: Relationship to the virus and host risk factors. J. Gastroenterol. Hepatol. 2006, 21, 1236–1239. [Google Scholar] [CrossRef] [PubMed]
- Macías, J.; Berenguer, J.; Japón, M.A.; Girón, J.A.; Rivero, A.; López-Cortés, L.F.; Moreno, A.; González-Serrano, M.; Iribarren, J.A.; Ortega, E.; et al. Fast fibrosis progression between repeated liver biopsies in patients coinfected with human immunodeficiency virus/hepatitis C virus. Hepatology 2009, 50, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Taura, N.; Ichikawa, T.; Hamasaki, K.; Nakao, K.; Nishimura, D.; Goto, T.; Fukuta, M.; Kawashimo, H.; Fujimoto, M.; Kusumoto, K.; et al. Association between liver fibrosis and insulin sensitivity in chronic hepatitis C patients. Am. J. Gastroenterol. 2006, 101, 2752–2759. [Google Scholar] [CrossRef] [PubMed]
- Fartoux, L.; Poujol-Robert, A.; Guéchot, J.; Wendum, D.; Poupon, R.; Serfaty, L. Insulin resistance is a cause of steatosis and fibrosis progression in chronic hepatitis C. Gut 2005, 54, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Perumalswami, P.; Kleiner, D.E.; Lutchman, G.; Heller, T.; Borg, B.; Park, Y.; Liang, T.J.; Hoofnagle, J.H.; Ghany, M.G. Steatosis and progression of fibrosis in untreated patients with chronic hepatitis C infection. Hepatology 2006, 43, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Moucari, R.; Asselah, T.; Cazals-Hatem, D.; Voitot, H.; Boyer, N.; Ripault, M.P.; Sobesky, R.; Martinot-Peignoux, M.; Maylin, S.; Nicolas-Chanoine, M.H.; et al. Insulin resistance in chronic hepatitis C: Association with genotypes 1 and 4, serum HCV RNA level, and liver fibrosis. Gastroenterology 2008, 134, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.X.; Kyulo, N.L.; Xia, V.W.; Hillebrand, D.J.; Hu, K.Q. Factors associated with hepatic fibrosis in patients with chronic hepatitis C: A retrospective study of a large cohort of U.S. patients. J. Clin. Gastroenterol. 2009, 43, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Paradis, V.; Perlemuter, G.; Bonvoust, F.; Dargere, D.; Parfait, B.; Vidaud, M.; Conti, M.; Huet, S.; Ba, N.; Buffet, C.; et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: A potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology 2001, 34, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Munteanu, M.; Charlotte, F.; Bonyhay, L.; Poynard, T. LIDO Study Group. Fibrogenic impact of high serum glucose in chronic hepatitis C. J. Hepatol. 2003, 39, 1049–1055. [Google Scholar] [CrossRef]
- Fartoux, L.; Chazouillères, O.; Wendum, D.; Poupon, R.; Serfaty, L. Impact of steatosis on progression of fibrosis in patients with mild hepatitis C. Hepatology 2005, 41, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.R.; Ilyas, J.; Duan, Z.; El-Serag, H.B. HCV genotype 3 is associated with an increased risk of cirrhosis and hepatocellular cancer in a national sample of U.S. Veterans with HCV. Hepatology 2014, 60, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Nkontchou, G.; Ziol, M.; Aout, M.; Lhabadie, M.; Baazia, Y.; Mahmoudi, A.; Roulot, D.; Ganne-Carrie, N.; Grando-Lemaire, V.; Trinchet, J.C.; et al. HCV genotype 3 is associated with a higher hepatocellular carcinoma incidence in patients with ongoing viral C cirrhosis. J. Viral Hepat. 2011, 18, e516–e522. [Google Scholar] [CrossRef] [PubMed]
- van der Meer, A.J.; Veldt, B.J.; Feld, J.J.; Wedemeyer, H.; Dufour, J.F.; Lammert, F.; Duarte-Rojo, A.; Heathcote, E.J.; Manns, M.P.; Kuske, L.; et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. JAMA 2012, 308, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Asahina, Y.; Tsuchiya, K.; Nishimura, T.; Muraoka, M.; Suzuki, Y.; Tamaki, N.; Yasui, Y.; Hosokawa, T.; Ueda, K.; Nakanishi, H.; et al. α-fetoprotein levels after interferon therapy and risk of hepatocarcinogenesis in chronic hepatitis C. Hepatology 2013, 58, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Salomao, M.; Yu, W.M.; Brown, R.S.; Emond, J.C.; Lefkowitch, J.H. Steatohepatitic hepatocellular carcinoma (SH-HCC): A distinctive histological variant of HCC in hepatitis C virus-related cirrhosis with associated NAFLD/NASH. Am. J. Surg. Pathol. 2010, 34, 1630–1636. [Google Scholar] [CrossRef] [PubMed]
- Nojiri, K.; Sugimoto, K.; Shiraki, K.; Kusagawa, S.; Tanaka, J.; Beppu, T.; Yamamoto, N.; Takei, Y.; Hashimoto, A.; Shimizu, A.; et al. Development of hepatocellular carcinoma in patients with chronic hepatitis C more than 10 years after sustained virological response to interferon therapy. Oncol. Lett. 2010, 1, 427–430. [Google Scholar] [PubMed]
- Tanaka, A.; Uegaki, S.; Kurihara, H.; Aida, K.; Mikami, M.; Nagashima, I.; Shiga, J.; Takikawa, H. Hepatic steatosis as a possible risk factor for the development of hepatocellular carcinoma after eradication of hepatitis C virus with antiviral therapy in patients with chronic hepatitis C. World J. Gastroenterol. 2007, 13, 5180–5187. [Google Scholar] [CrossRef] [PubMed]
- Takuma, Y.; Nouso, K.; Makino, Y.; Saito, S.; Takayama, H.; Takahara, M.; Takahashi, H.; Murakami, I.; Takeuchi, H. Hepatic steatosis correlates with the postoperative recurrence of hepatitis C virus-associated hepatocellular carcinoma. Liver Int. 2007, 27, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Pekow, J.R.; Bhan, A.K.; Zheng, H.; Chung, R.T. Hepatic steatosis is associated with increased frequency of hepatocellular carcinoma in patients with hepatitis C-related cirrhosis. Cancer 2007, 109, 2490–2496. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Farrell, G.C.; Kench, J.; George, J. Hepatic steatosis and the risk of hepatocellular carcinoma in chronic hepatitis C. J. Gastroenterol. Hepatol. 2005, 20, 1395–1400. [Google Scholar] [CrossRef] [PubMed]
- Ohata, K.; Hamasaki, K.; Toriyama, K.; Matsumoto, K.; Saeki, A.; Yanagi, K.; Abiru, S.; Nakagawa, Y.; Shigeno, M.; Miyazoe, S.; et al. Hepatic steatosis is a risk factor for hepatocellular carcinoma in patients with chronic hepatitis C virus infection. Cancer 2003, 97, 3036–3043. [Google Scholar] [CrossRef] [PubMed]
- Tazawa, J.; Maeda, M.; Nakagawa, M.; Ohbayashi, H.; Kusano, F.; Yamane, M.; Sakai, Y.; Suzuki, K. Diabetes mellitus may be associated with hepatocarcinogenesis in patients with chronic hepatitis C. Dig. Dis. Sci. 2002, 47, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Jahan, S.; Ashfaq, U.A.; Qasim, M.; Khaliq, S.; Saleem, M.J.; Afzal, N. Hepatitis C virus to hepatocellular carcinoma. Infect. Agents Cancer 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Koike, K. Hepatitis C virus contributes to hepatocarcinogenesis by modulating metabolic and intracellular signaling pathways. J. Gastroenterol. Hepatol. 2007, 22, S108–S111. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, L.E.; Restivo, L.; Zampino, R.; Lonardo, A.; Loria, P. Metabolic alterations and chronic hepatitis C: Treatment strategies. Expert Opin. Pharmacother. 2011, 12, 2215–2234. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.H.; Brancati, F.L.; Sulkowski, M.S.; Strathdee, S.A.; Szklo, M.; Thomas, D.L. Prevalence of type 2 diabetes mellitus among persons with hepatitis C virus infection in the United States. Ann. Intern. Med. 2000, 133, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Simó, R.; Hernández, C.; Genescà, J.; Jardí, R.; Mesa, J. High prevalence of hepatitis C virus infection in diabetic patients. Diabetes Care 1996, 19, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Hadziyannis, S.J. The spectrum of extrahepatic manifestations in hepatitis C virus infection. J. Viral Hepat. 1997, 4, 9–28. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Cammà, C.; di Marco, V.; Alessi, N.; Cabibi, D.; Caldarella, R.; Licata, A.; Massenti, F.; Tarantino, G.; Marchesini, G.; et al. Insulin resistance and diabetes increase fibrosis in the liver of patients with genotype 1 HCV infection. Am. J. Gastroenterol. 2008, 103, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.Y.; Kim, S.S.; Kwon, O.S.; Kwon, K.A.; Chung, M.G.; Park, D.K.; Kim, Y.S.; Koo, Y.S.; Kim, Y.K.; Choi, D.J.; et al. Prognostic significance of glycaemic control in patients with HBV and HCV-related cirrhosis and diabetes mellitus. Diabet. Med. 2005, 22, 1530–1535. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Yang, H.I.; Yang, W.S.; Liu, C.J.; Chen, P.J.; You, S.L.; Wang, L.Y.; Sun, C.A.; Lu, S.N.; Chen, D.S.; et al. Metabolic factors and risk of hepatocellular carcinoma by chronic hepatitis B/C infection: A follow-up study in Taiwan. Gastroenterology 2008, 135, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Boddi, M.; Abbate, R.; Chellini, B.; Giusti, B.; Giannini, C.; Pratesi, G.; Rossi, L.; Pratesi, C.; Gensini, G.F.; Paperetti, L.; et al. Hepatitis C virus RNA localization in human carotid plaques. J. Clin. Virol. 2010, 47, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Ampuero, J.; Romero-Gómez, M.; Reddy, K.R. HCV genotype 3. The new treatment challenge. Aliment. Pharmacol. Ther. 2014, 39, 686–698. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adinolfi, L.E.; Rinaldi, L.; Guerrera, B.; Restivo, L.; Marrone, A.; Giordano, M.; Zampino, R. NAFLD and NASH in HCV Infection: Prevalence and Significance in Hepatic and Extrahepatic Manifestations. Int. J. Mol. Sci. 2016, 17, 803. https://doi.org/10.3390/ijms17060803
Adinolfi LE, Rinaldi L, Guerrera B, Restivo L, Marrone A, Giordano M, Zampino R. NAFLD and NASH in HCV Infection: Prevalence and Significance in Hepatic and Extrahepatic Manifestations. International Journal of Molecular Sciences. 2016; 17(6):803. https://doi.org/10.3390/ijms17060803
Chicago/Turabian StyleAdinolfi, Luigi Elio, Luca Rinaldi, Barbara Guerrera, Luciano Restivo, Aldo Marrone, Mauro Giordano, and Rosa Zampino. 2016. "NAFLD and NASH in HCV Infection: Prevalence and Significance in Hepatic and Extrahepatic Manifestations" International Journal of Molecular Sciences 17, no. 6: 803. https://doi.org/10.3390/ijms17060803
APA StyleAdinolfi, L. E., Rinaldi, L., Guerrera, B., Restivo, L., Marrone, A., Giordano, M., & Zampino, R. (2016). NAFLD and NASH in HCV Infection: Prevalence and Significance in Hepatic and Extrahepatic Manifestations. International Journal of Molecular Sciences, 17(6), 803. https://doi.org/10.3390/ijms17060803