
Role of Hedgehog Signaling Pathway in NASH

{kind=link}
{kind=link}
Abstract
:1. Introduction
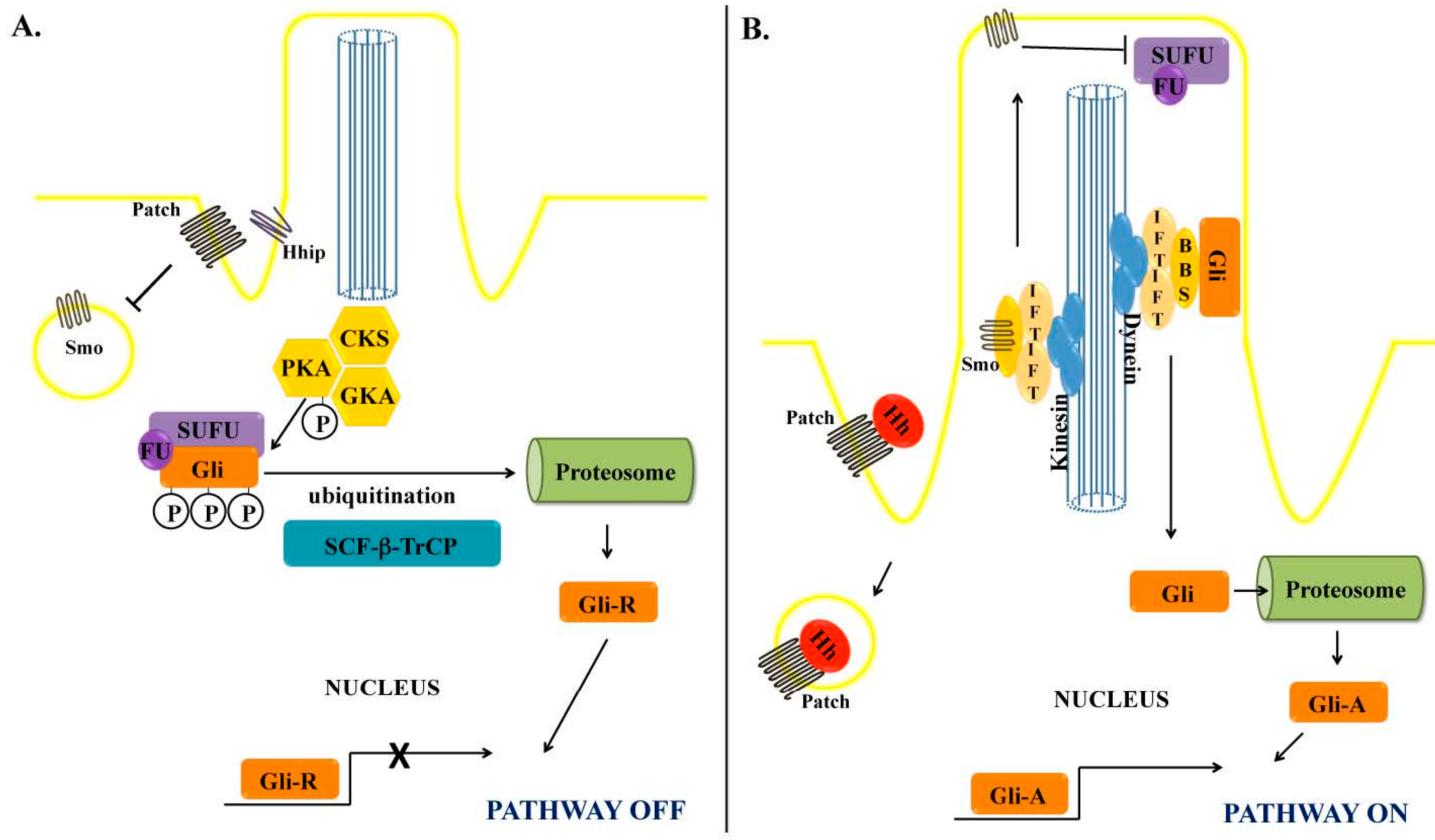
2. The Hedgehog Signaling Pathway
3. Hedgehog Pathway and the Wound Healing Response
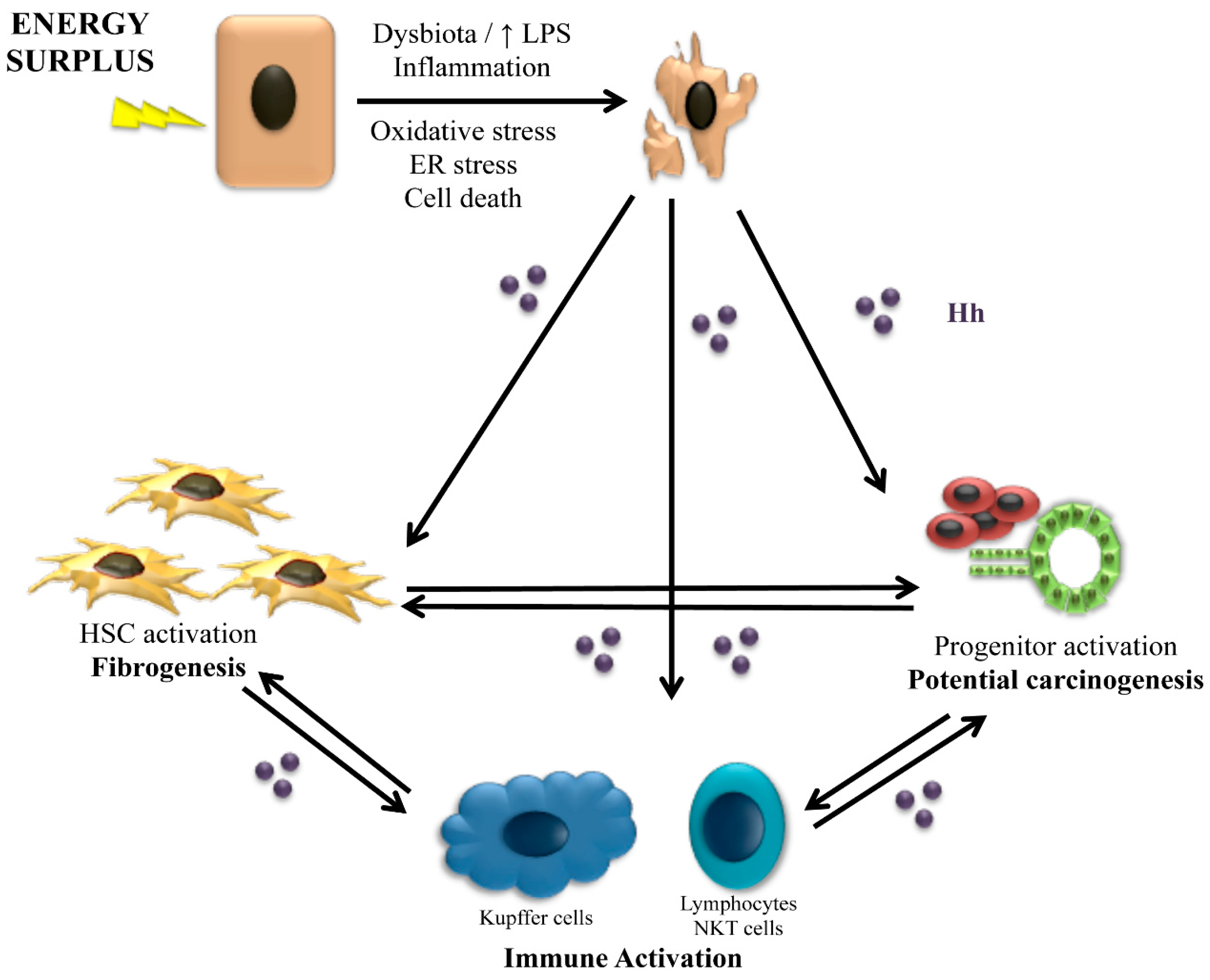
4. The Role of Hedgehog in Animal Models of NASH
5. The Hegdehog Pathway in Human NASH
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BBS | Bardet-Biedl syndrome proteins |
| Boc | brother of Cdo |
| Cdo | CAM-related downregulated by oncogenes |
| Cos | Costal-2 |
| CK1 | casein kinase-1 |
| Dhh | Desert hedgehog |
| Fu | fused kinase |
| GAS-1 | growth arrest-specific-1 |
| GPCR | G-protein-coupled receptor |
| GSK | glycogen synthase kinase |
| Hh | hedgehog |
| Hip | hedgehog-interacting protein |
| HSC | hepatic stellate cell |
| IFP | intraflagellar transport proteins |
| Ihh | Indian hedgehog |
| IL | interleukin |
| MBOAT | membrane-bound O-acyltransferase |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
| PDGF | platelet-derived growth factor |
| PKA | protein kinase A |
| Ptch | Ptched |
| Shh | Sonic hedgehog |
| SKI | skinny hedgehog |
| SMA | smooth muscle actin |
| Smo | smoothened |
| Sufu | suppressor of fused |
| TGF | transforming growth factor |
| TrCp | transducing repeat-containing protein |
| VEGF | vascular endothelial growth factor |
| VLDL | very low-density lipoproteins |
References
- Loomba, R.; Sanyal, A.J. The global nafld epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Bellentani, S.; Argo, C.K.; Ballestri, S.; Byrne, C.D.; Caldwell, S.H.; Cortez-Pinto, H.; Grieco, A.; Machado, M.V.; Miele, L.; et al. Epidemiological modifiers of non-alcoholic fatty liver disease: Focus on high-risk groups. Dig. Liver Dis. 2015, 47, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Diehl, A.M. Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology 2006, 40, S17–S29. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Cortez-Pinto, H. Non-alcoholic fatty liver disease: What the clinician needs to know. World J. Gastroenterol. 2014, 20, 12956–12980. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the united states. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Cheung, R.; Ahmed, A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 2014, 59, 2188–2195. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015, 149, 389–397.e10. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Hagstrom, H.; Nasr, P.; Fredrikson, M.; Stal, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in nafld after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654, e641–649; quiz e639–640. [Google Scholar] [CrossRef] [PubMed]
- Moylan, C.A.; Pang, H.; Dellinger, A.; Suzuki, A.; Garrett, M.E.; Guy, C.D.; Murphy, S.K.; Ashley-Koch, A.E.; Choi, S.S.; Michelotti, G.A.; et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014, 59, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Horn, A.; Palumbo, K.; Cordazzo, C.; Dees, C.; Akhmetshina, A.; Tomcik, M.; Zerr, P.; Avouac, J.; Gusinde, J.; Zwerina, J.; et al. Hedgehog signaling controls fibroblast activation and tissue fibrosis in systemic sclerosis. Arthritis Rheum. 2012, 64, 2724–2733. [Google Scholar] [CrossRef] [PubMed]
- Kugler, M.C.; Joyner, A.L.; Loomis, C.A.; Munger, J.S. Sonic hedgehog signaling in the lung. From development to disease. Am. J. Respir. Cell Mol. Biol. 2015, 52, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fabian, S.L.; Penchev, R.R.; St-Jacques, B.; Rao, A.N.; Sipila, P.; West, K.A.; McMahon, A.P.; Humphreys, B.D. Hedgehog-gli pathway activation during kidney fibrosis. Am. J. Pathol. 2012, 180, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.W.; Lin, H.; Lu, Y.; Xia, W.; Gao, J.; Li, Z.S. Sonic hedgehog expression in a rat model of chronic pancreatitis. World J. Gastroenterol. 2014, 20, 4712–4717. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Omenetti, A.; Syn, W.K.; Diehl, A.M. The role of hedgehog signaling in fibrogenic liver repair. Int. J. Biochem. Cell Biol. 2011, 43, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Therond, P.P. The mechanisms of hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Farzan, S.F.; Singh, S.; Schilling, N.S.; Robbins, D.J. The adventures of sonic hedgehog in development and repair. III. Hedgehog processing and biological activity. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G844–G849. [Google Scholar] [CrossRef] [PubMed]
- Pathi, S.; Pagan-Westphal, S.; Baker, D.P.; Garber, E.A.; Rayhorn, P.; Bumcrot, D.; Tabin, C.J.; Blake Pepinsky, R.; Williams, K.P. Comparative biological responses to human sonic, indian, and desert hedgehog. Mech. Dev. 2001, 106, 107–117. [Google Scholar] [CrossRef]
- Merchant, J.L.; Saqui-Salces, M. Inhibition of hedgehog signaling in the gastrointestinal tract: Targeting the cancer microenvironment. Cancer Treat. Rev. 2014, 40, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Lin, X.; Lu, H.; Chen, B.; Bai, Y. An overview of hedgehog signaling in fibrosis. Mol. Pharmacol. 2015, 87, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Teperino, R.; Aberger, F.; Esterbauer, H.; Riobo, N.; Pospisilik, J.A. Canonical and non-canonical hedgehog signalling and the control of metabolism. Semin. Cell Dev. Biol. 2014, 33, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Kolterud, A.; Zeng, H.; Hoover, A.; Teglund, S.; Toftgard, R.; Liu, A. Suppressor of fused inhibits mammalian hedgehog signaling in the absence of cilia. Dev. Biol. 2009, 330, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.S.; Neill, G.W.; Regl, G.; Eichberger, T.; Frischauf, A.M.; Aberger, F.; Quinn, A.; Philpott, M. Gli2 is expressed in normal human epidermis and bcc and induces Gli1 expression by binding to its promoter. J. Investig. Dermatol. 2004, 122, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- Roy, S. Cilia and hedgehog: When and how was their marriage solemnized? Differentiation 2012, 83, S43–S48. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.A.; Nishi, Y.; Ma, W.; Vedenko, A.; Shokri, L.; Zhang, X.; McFarlane, M.; Baizabal, J.M.; Junker, J.P.; van Oudenaarden, A.; et al. Neural-specific Sox2 input and differential Gli-binding affinity provide context and positional information in Shh-directed neural patterning. Genes Dev. 2012, 26, 2802–2816. [Google Scholar] [CrossRef] [PubMed]
- Chinchilla, P.; Xiao, L.; Kazanietz, M.G.; Riobo, N.A. Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle 2010, 9, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Pasca di Magliano, M.; Hebrok, M. Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 2003, 3, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E.A.; Kong, M.; Ollendorff, V.; Donoghue, D.J. Patched1 interacts with cyclin B1 to regulate cell cycle progression. EMBO J. 2001, 20, 2214–2223. [Google Scholar] [CrossRef] [PubMed]
- Polizio, A.H.; Chinchilla, P.; Chen, X.; Manning, D.R.; Riobo, N.A. Sonic hedgehog activates the gtpases rac1 and rhoa in a Gli-independent manner through coupling of smoothened to Gi proteins. Sci. Signal. 2011, 4 Pt 7. [Google Scholar] [CrossRef] [PubMed]
- Teperino, R.; Amann, S.; Bayer, M.; McGee, S.L.; Loipetzberger, A.; Connor, T.; Jaeger, C.; Kammerer, B.; Winter, L.; Wiche, G.; et al. Hedgehog partial agonism drives warburg-like metabolism in muscle and brown fat. Cell 2012, 151, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, M.F.; Borensztajn, K.S.; Roelink, H.; Peppelenbosch, M.P.; Spek, C.A. Sonic hedgehog induces transcription-independent cytoskeletal rearrangement and migration regulated by arachidonate metabolites. Cell Signal. 2007, 19, 2596–2604. [Google Scholar] [CrossRef] [PubMed]
- Polizio, A.H.; Chinchilla, P.; Chen, X.; Kim, S.; Manning, D.R.; Riobo, N.A. Heterotrimeric Gi proteins link hedgehog signaling to activation of rho small gtpases to promote fibroblast migration. J. Biol. Chem. 2011, 286, 19589–19596. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gradilone, S.A.; Smoot, R.L.; Mertens, J.C.; Bronk, S.F.; Sirica, A.E.; Gores, G.J. Non-canonical hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. J. Hepatol. 2014, 60, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Samant, R.S.; Shevde, L.A. Nonclassical activation of hedgehog signaling enhances multidrug resistance and makes cancer cells refractory to smoothened-targeting hedgehog inhibition. J. Biol. Chem. 2013, 288, 11824–11833. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, G.A.; Xie, G.; Swiderska, M.; Choi, S.S.; Karaca, G.; Kruger, L.; Premont, R.; Yang, L.; Syn, W.K.; Metzger, D.; et al. Smoothened is a master regulator of adult liver repair. J. Clin. Investig. 2013, 123, 2380–2394. [Google Scholar] [CrossRef] [PubMed]
- Sommerfeld, A.; Reinehr, R.; Haussinger, D. Free fatty acids shift insulin-induced hepatocyte proliferation towards CD95-dependent apoptosis. J. Biol. Chem. 2015, 290, 4398–4409. [Google Scholar] [CrossRef] [PubMed]
- Mooney, C.J.; Hakimjavadi, R.; Fitzpatrick, E.; Kennedy, E.; Walls, D.; Morrow, D.; Redmond, E.M.; Cahill, P.A. Hedgehog and resident vascular stem cell fate. Stem Cells Int. 2015, 2015, 468428. [Google Scholar] [CrossRef] [PubMed]
- Sicklick, J.K.; Li, Y.X.; Melhem, A.; Schmelzer, E.; Zdanowicz, M.; Huang, J.; Caballero, M.; Fair, J.H.; Ludlow, J.W.; McClelland, R.E.; et al. Hedgehog signaling maintains resident hepatic progenitors throughout life. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G859–G870. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Witek, R.P.; Syn, W.K.; Choi, S.S.; Omenetti, A.; Premont, R.; Guy, C.D.; Diehl, A.M. Signals from dying hepatocytes trigger growth of liver progenitors. Gut 2010, 59, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Fleig, S.V.; Choi, S.S.; Yang, L.; Jung, Y.; Omenetti, A.; VanDongen, H.M.; Huang, J.; Sicklick, J.K.; Diehl, A.M. Hepatic accumulation of hedgehog-reactive progenitors increases with severity of fatty liver damage in mice. Lab. Investig. 2007, 87, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Brown, K.D.; Witek, R.P.; Omenetti, A.; Yang, L.; Vandongen, M.; Milton, R.J.; Hines, I.N.; Rippe, R.A.; Spahr, L.; et al. Accumulation of hedgehog-responsive progenitors parallels alcoholic liver disease severity in mice and humans. Gastroenterology 2008, 134, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Itoh, T.; Miyajima, A. Hedgehog signal activation coordinates proliferation and differentiation of fetal liver progenitor cells. Exp. Cell Res. 2009, 315, 2648–2657. [Google Scholar] [CrossRef] [PubMed]
- Syn, W.K.; Witek, R.P.; Curbishley, S.M.; Jung, Y.; Choi, S.S.; Enrich, B.; Omenetti, A.; Agboola, K.M.; Fearing, C.M.; Tilg, H.; et al. Role for hedgehog pathway in regulating growth and function of invariant NKT cells. Eur. J. Immunol. 2009, 39, 1879–1892. [Google Scholar] [CrossRef] [PubMed]
- Pereira, T.A.; Xie, G.; Choi, S.S.; Syn, W.K.; Voieta, I.; Lu, J.; Chan, I.S.; Swiderska, M.; Amaral, K.B.; Antunes, C.M.; et al. Macrophage-derived hedgehog ligands promotes fibrogenic and angiogenic responses in human schistosomiasis mansoni. Liver Int. 2013, 33, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Iwaisako, K.; Brenner, D.A.; Kisseleva, T. What’s new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. J. Gastroenterol. Hepatol. 2012, 27 (Suppl. 2), 65–68. [Google Scholar] [CrossRef] [PubMed]
- Swiderska-Syn, M.; Syn, W.K.; Xie, G.; Kruger, L.; Machado, M.V.; Karaca, G.; Michelotti, G.A.; Choi, S.S.; Premont, R.T.; Diehl, A.M. Myofibroblastic cells function as progenitors to regenerate murine livers after partial hepatectomy. Gut 2013. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, Y.; Mao, H.; Fleig, S.; Omenetti, A.; Brown, K.D.; Sicklick, J.K.; Li, Y.X.; Diehl, A.M. Sonic hedgehog is an autocrine viability factor for myofibroblastic hepatic stellate cells. J. Hepatol. 2008, 48, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Choi, S.S.; Syn, W.K.; Michelotti, G.A.; Swiderska, M.; Karaca, G.; Chan, I.S.; Chen, Y.; Diehl, A.M. Hedgehog signalling regulates liver sinusoidal endothelial cell capillarisation. Gut 2013, 62, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Guy, C.D.; Suzuki, A.; Zdanowicz, M.; Abdelmalek, M.F.; Burchette, J.; Unalp, A.; Diehl, A.M.; Nash, C.R.N. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012, 55, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, F.; Guy, C.D.; Lu, J.; Suzuki, A.; Burchette, J.L.; Abdelmalek, M.F.; Chen, W.; Diehl, A.M. Increased production of sonic hedgehog by ballooned hepatocytes. J. Pathol. 2011, 224, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Michelotti, G.A.; Pereira Tde, A.; Boursier, J.; Kruger, L.; Swiderska-Syn, M.; Karaca, G.; Xie, G.; Guy, C.D.; Bohinc, B.; et al. Reduced lipoapoptosis, hedgehog pathway activation and fibrosis in caspase-2 deficient mice with non-alcoholic steatohepatitis. Gut 2015, 64, 1148–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omenetti, A.; Porrello, A.; Jung, Y.; Yang, L.; Popov, Y.; Choi, S.S.; Witek, R.P.; Alpini, G.; Venter, J.; Vandongen, H.M.; et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J. Clin. Investig. 2008, 118, 3331–3342. [Google Scholar] [CrossRef] [PubMed]
- Sicklick, J.K.; Li, Y.X.; Choi, S.S.; Qi, Y.; Chen, W.; Bustamante, M.; Huang, J.; Zdanowicz, M.; Camp, T.; Torbenson, M.S.; et al. Role for hedgehog signaling in hepatic stellate cell activation and viability. Lab. Investig. 2005, 85, 1368–1380. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Tang, Z.; Deng, M.; Zhong, Y.; Lin, J.; Yang, X.; Xiang, P.; Xu, R. Hedgehog-mediated paracrine interaction between hepatic stellate cells and marrow-derived mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2008, 372, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Frank, D.B.; Kadzik, R.S.; Morley, M.P.; Rathi, K.S.; Wang, T.; Zhou, S.; Cheng, L.; Lu, M.M.; Morrisey, E.E. Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration. Nature 2015, 526, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Popov, Y.; Jung, Y.; Choi, S.S.; Witek, R.P.; Yang, L.; Brown, K.D.; Schuppan, D.; Diehl, A.M. The hedgehog pathway regulates remodelling responses to biliary obstruction in rats. Gut 2008, 57, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Syn, W.K.; Jung, Y.; Francis, H.; Porrello, A.; Witek, R.P.; Choi, S.S.; Yang, L.; Mayo, M.J.; Gershwin, M.E.; et al. Repair-related activation of hedgehog signaling promotes cholangiocyte chemokine production. Hepatology 2009, 50, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Syn, W.K.; Jung, Y.; Omenetti, A.; Abdelmalek, M.; Guy, C.D.; Yang, L.; Wang, J.; Witek, R.P.; Fearing, C.M.; Pereira, T.A.; et al. Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology 2009, 137, 1478–1488.e8. [Google Scholar] [CrossRef] [PubMed]
- Syn, W.K.; Oo, Y.H.; Pereira, T.A.; Karaca, G.F.; Jung, Y.; Omenetti, A.; Witek, R.P.; Choi, S.S.; Guy, C.D.; Fearing, C.M.; et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 2010, 51, 1998–2007. [Google Scholar] [CrossRef] [PubMed]
- Syn, W.K.; Choi, S.S.; Liaskou, E.; Karaca, G.F.; Agboola, K.M.; Oo, Y.H.; Mi, Z.; Pereira, T.A.; Zdanowicz, M.; Malladi, P.; et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 2011, 53, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Pazzaglia, S.; Cifaldi, L.; Saran, A.; Nobili, V.; Fruci, D.; Alisi, A. Hedgehog/hyaluronic acid interaction network in nonalcoholic fatty liver disease, fibrosis, and hepatocellular carcinoma. Hepatology 2012, 56, 1589. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; Almeida Pereira, T.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakisaka, K.; Cazanave, S.C.; Werneburg, N.W.; Razumilava, N.; Mertens, J.C.; Bronk, S.F.; Gores, G.J. A hedgehog survival pathway in ‘undead’ lipotoxic hepatocytes. J. Hepatol. 2012, 57, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, H.; Nakamura, M.; Yamaguchi, H.; Yamanaka, N.; Akiyoshi, T.; Koga, K.; Yamaguchi, K.; Tsuneyoshi, M.; Tanaka, M.; Katano, M. Nuclear factor-kappab contributes to hedgehog signaling pathway activation through sonic hedgehog induction in pancreatic cancer. Cancer Res. 2006, 66, 7041–7049. [Google Scholar] [CrossRef] [PubMed]
- Syn, W.K.; Agboola, K.M.; Swiderska, M.; Michelotti, G.A.; Liaskou, E.; Pang, H.; Xie, G.; Philips, G.; Chan, I.S.; Karaca, G.F.; et al. Nkt-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut 2012, 61, 1323–1329. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Song, K.; Han, C.; Chen, W.; Wang, Y.; Dash, S.; Lim, K.; Wu, T. Inhibition of hedgehog signaling ameliorates hepatic inflammation in mice with nonalcoholic fatty liver disease. Hepatology 2015. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.I.; Moon, H.; Ju, H.L.; Cho, K.J.; Kim, D.Y.; Han, K.H.; Eun, J.W.; Nam, S.W.; Ribback, S.; Dombrowski, F.; et al. Hepatic expression of sonic hedgehog induces liver fibrosis and promotes hepatocarcinogenesis in a transgenic mouse model. J. Hepatol. 2015, 64, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Hirsova, P.; Ibrahim, S.H.; Bronk, S.F.; Yagita, H.; Gores, G.J. Vismodegib suppresses trail-mediated liver injury in a mouse model of nonalcoholic steatohepatitis. PLoS ONE 2013, 8, e70599. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Michelotti, G.A.; Pereira, T.A.; Xie, G.; Premont, R.; Cortez-Pinto, H.; Diehl, A.M. Accumulation of duct cells with activated yap parallels fibrosis progression in non-alcoholic fatty liver disease. J. Hepatol. 2015, 63, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.M.; Jonsson, J.R.; Powell, E.E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; Weltman, M.D.; Tilg, H.; Moschen, A.R.; et al. Progressive fibrosis in nonalcoholic steatohepatitis: Association with altered regeneration and a ductular reaction. Gastroenterology 2007, 133, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Jing, Y.Y.; Guo, S.W.; Yu, G.F.; Fan, Q.M.; Qu, F.F.; Gao, L.; Yang, Y.; Wu, D.; Meng, Y.; et al. Proliferative ductular reactions correlate with hepatic progenitor cell and predict recurrence in hcc patients after curative resection. Cell Biosci. 2014, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Swiderska-Syn, M.; Suzuki, A.; Guy, C.D.; Schwimmer, J.B.; Abdelmalek, M.F.; Lavine, J.E.; Diehl, A.M. Hedgehog pathway and pediatric nonalcoholic fatty liver disease. Hepatology 2013, 57, 1814–1825. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Bass, L.M.; Anders, R.A.; Clemente, M.G.; Francis, H.; Guy, C.D.; McCall, S.; Choi, S.S.; Alpini, G.; Schwarz, K.B.; et al. Hedgehog activity, epithelial-mesenchymal transitions, and biliary dysmorphogenesis in biliary atresia. Hepatology 2011, 53, 1246–1258. [Google Scholar] [CrossRef] [PubMed]
- Guy, C.D.; Suzuki, A.; Abdelmalek, M.F.; Burchette, J.L.; Diehl, A.M.; NASH CRN. Treatment response in the pivens trial is associated with decreased hedgehog pathway activity. Hepatology 2015, 61, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Grzelak, C.A.; Martelotto, L.G.; Sigglekow, N.D.; Patkunanathan, B.; Ajami, K.; Calabro, S.R.; Dwyer, B.J.; Tirnitz-Parker, J.E.; Watkins, D.N.; Warner, F.J.; et al. The intrahepatic signalling niche of hedgehog is defined by primary cilia positive cells during chronic liver injury. J. Hepatol. 2014, 60, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Matz-Soja, M.; Gebhardt, R. The many faces of hedgehog signalling in the liver: Recent progress reveals striking cellular diversity and the importance of microenvironments. J. Hepatol. 2014, 61, 1449–1450. [Google Scholar] [CrossRef] [PubMed]
- Uschner, F.E.; Ranabhat, G.; Choi, S.S.; Granzow, M.; Klein, S.; Schierwagen, R.; Raskopf, E.; Gautsch, S.; van der Ven, P.F.; Furst, D.O.; et al. Statins activate the canonical hedgehog-signaling and aggravate non-cirrhotic portal hypertension, but inhibit the non-canonical hedgehog signaling and cirrhotic portal hypertension. Sci. Rep. 2015, 5, 14573. [Google Scholar] [CrossRef] [PubMed]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.; Reiter, J.F. Vertebrate smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, B.; Syn, W.K.; Delgado, I.; Karaca, G.F.; Jung, Y.; Wang, J.; Zubiaga, A.M.; Fresnedo, O.; Omenetti, A.; Zdanowicz, M.; et al. Hedgehog signaling is critical for normal liver regeneration after partial hepatectomy in mice. Hepatology 2010, 51, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.C.; McGlashan, S.R.; Cooling, M.T.; Long, D.S. Culture and detection of primary cilia in endothelial cell models. Cilia 2015, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Guha, M. Hedgehog inhibitor gets landmark skin cancer approval, but questions remain for wider potential. Nat. Rev. Drug Discov. 2012, 11, 257–258. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verdelho Machado, M.; Diehl, A.M. Role of Hedgehog Signaling Pathway in NASH. Int. J. Mol. Sci. 2016, 17, 857. https://doi.org/10.3390/ijms17060857
Verdelho Machado M, Diehl AM. Role of Hedgehog Signaling Pathway in NASH. International Journal of Molecular Sciences. 2016; 17(6):857. https://doi.org/10.3390/ijms17060857
Chicago/Turabian StyleVerdelho Machado, Mariana, and Anna Mae Diehl. 2016. "Role of Hedgehog Signaling Pathway in NASH" International Journal of Molecular Sciences 17, no. 6: 857. https://doi.org/10.3390/ijms17060857
APA StyleVerdelho Machado, M., & Diehl, A. M. (2016). Role of Hedgehog Signaling Pathway in NASH. International Journal of Molecular Sciences, 17(6), 857. https://doi.org/10.3390/ijms17060857