
Comparative Mitogenomic Analysis of Species Representing Six Subfamilies in the Family Tenebrionidae

Abstract
:
1. Introduction
2. Results
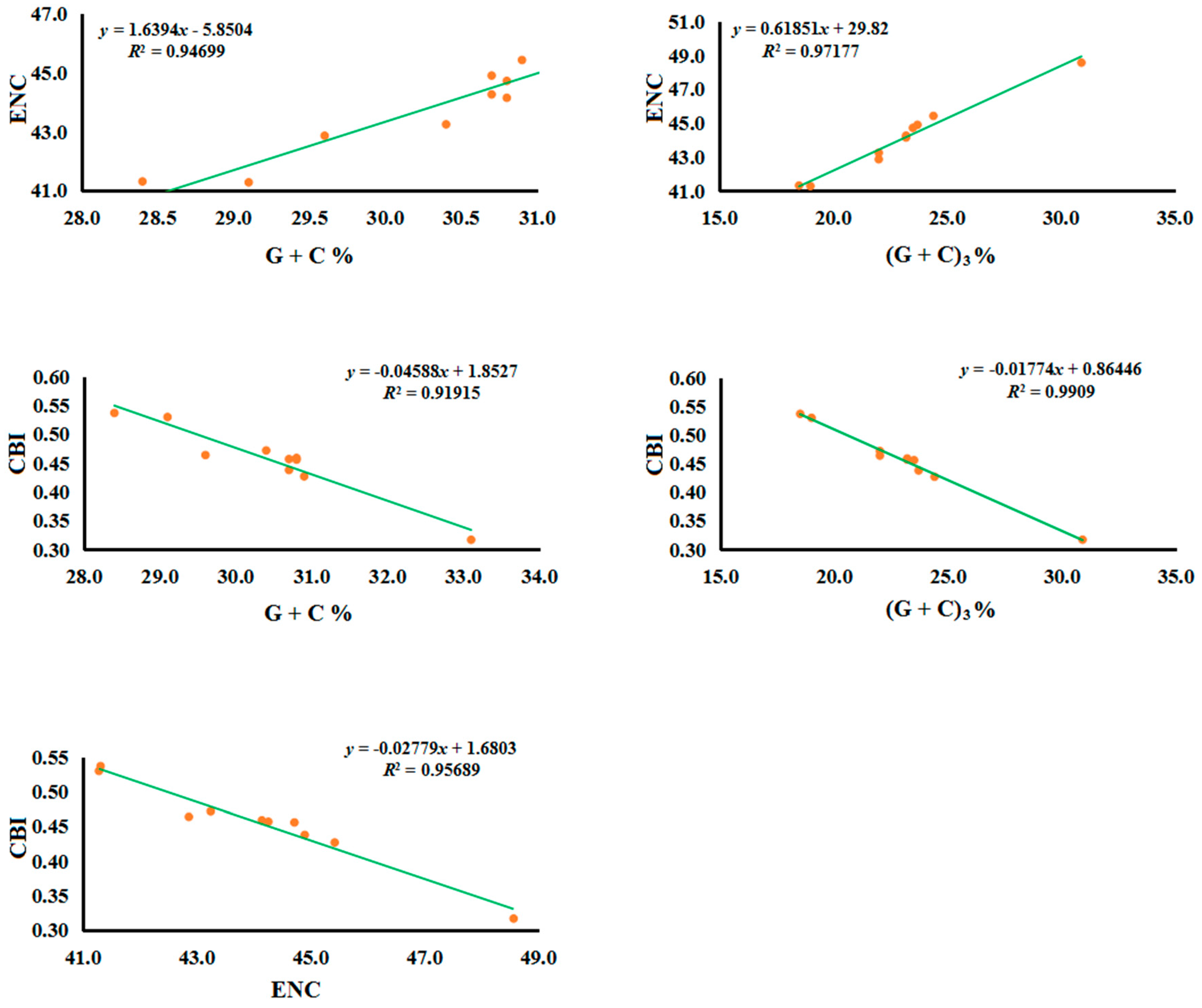
2.1. Nucleotide Composition
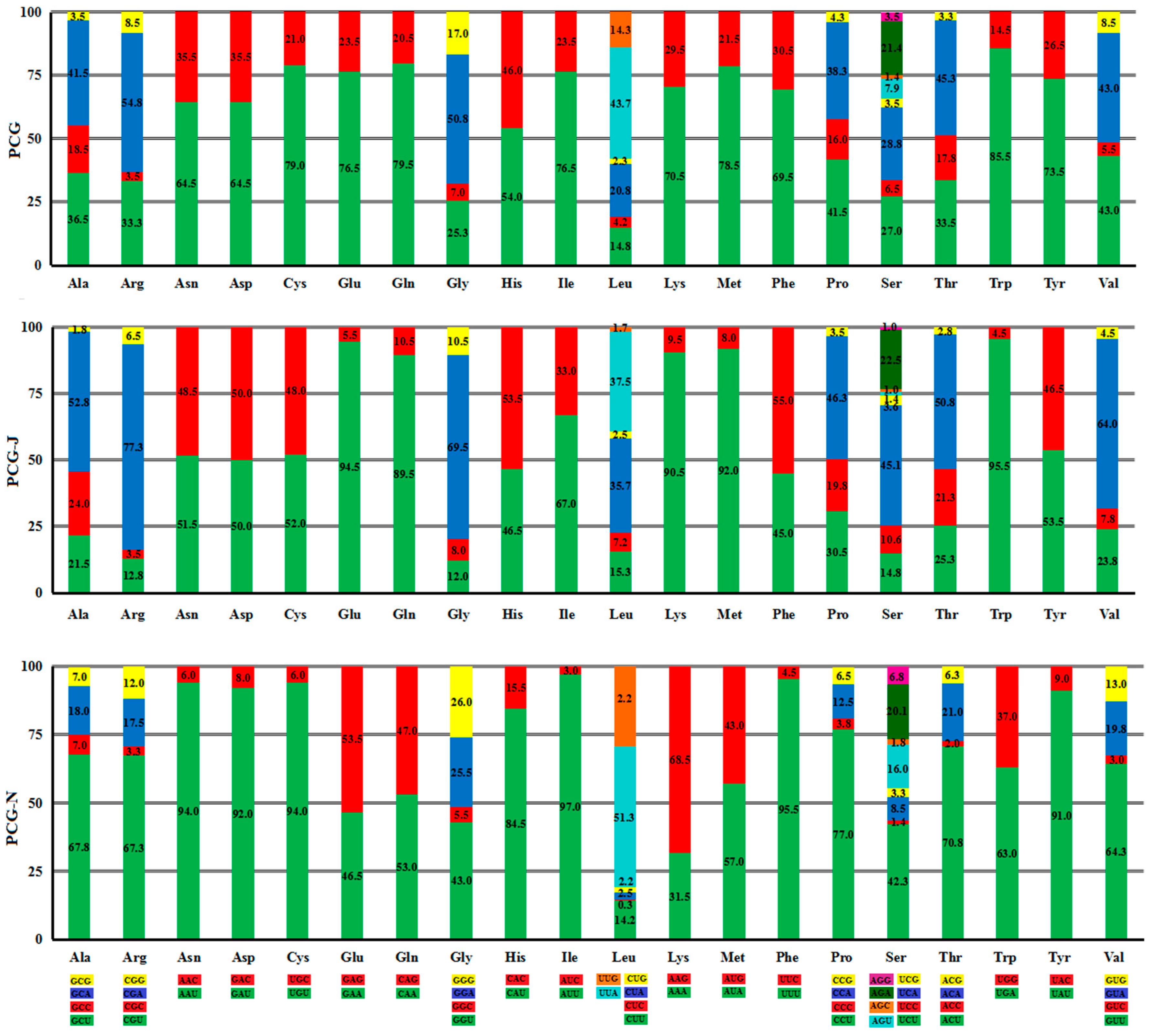
2.2. Protein-Coding Genes (PCGs)
2.2.1. Initiation and Termination Codons
2.2.2. Codon Usage
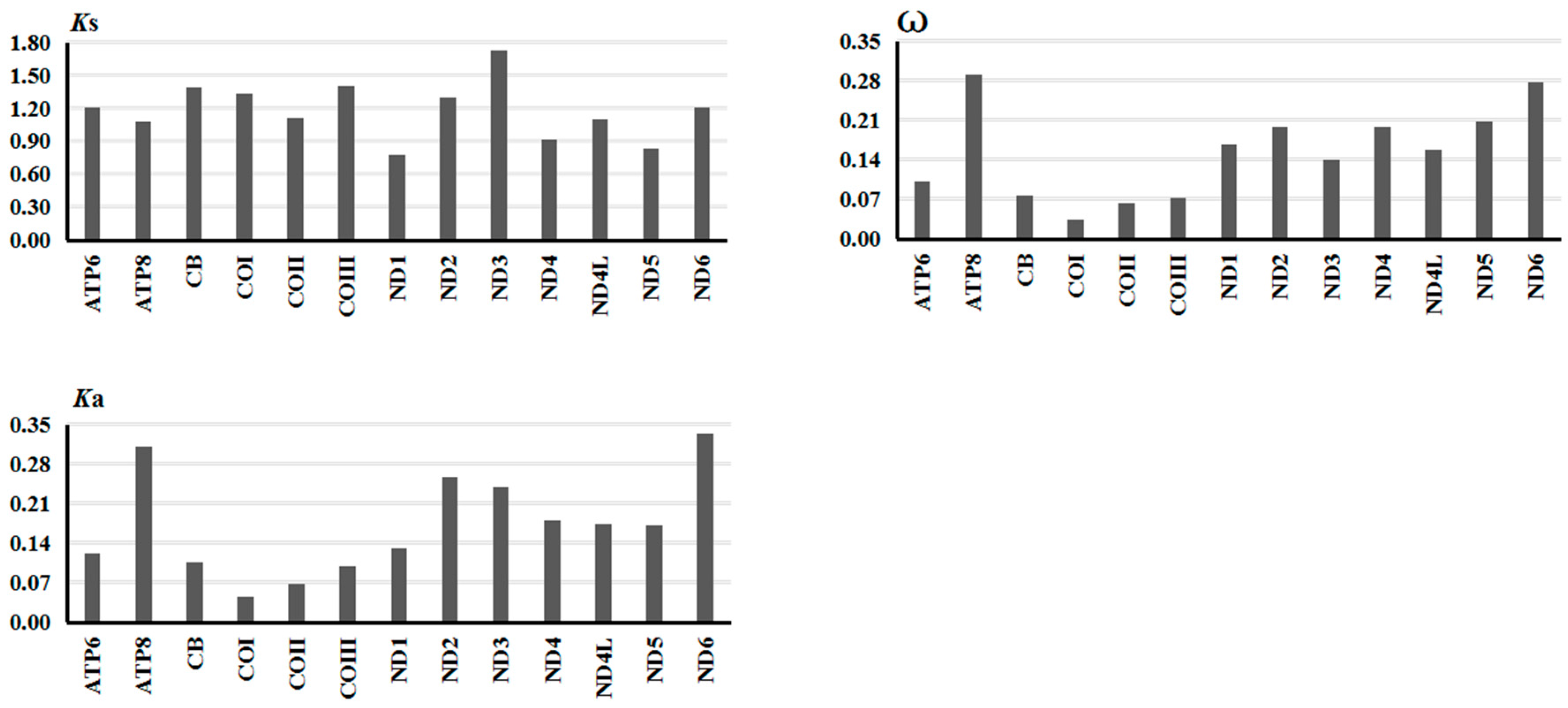
2.2.3. Gene Evolutionary Rate
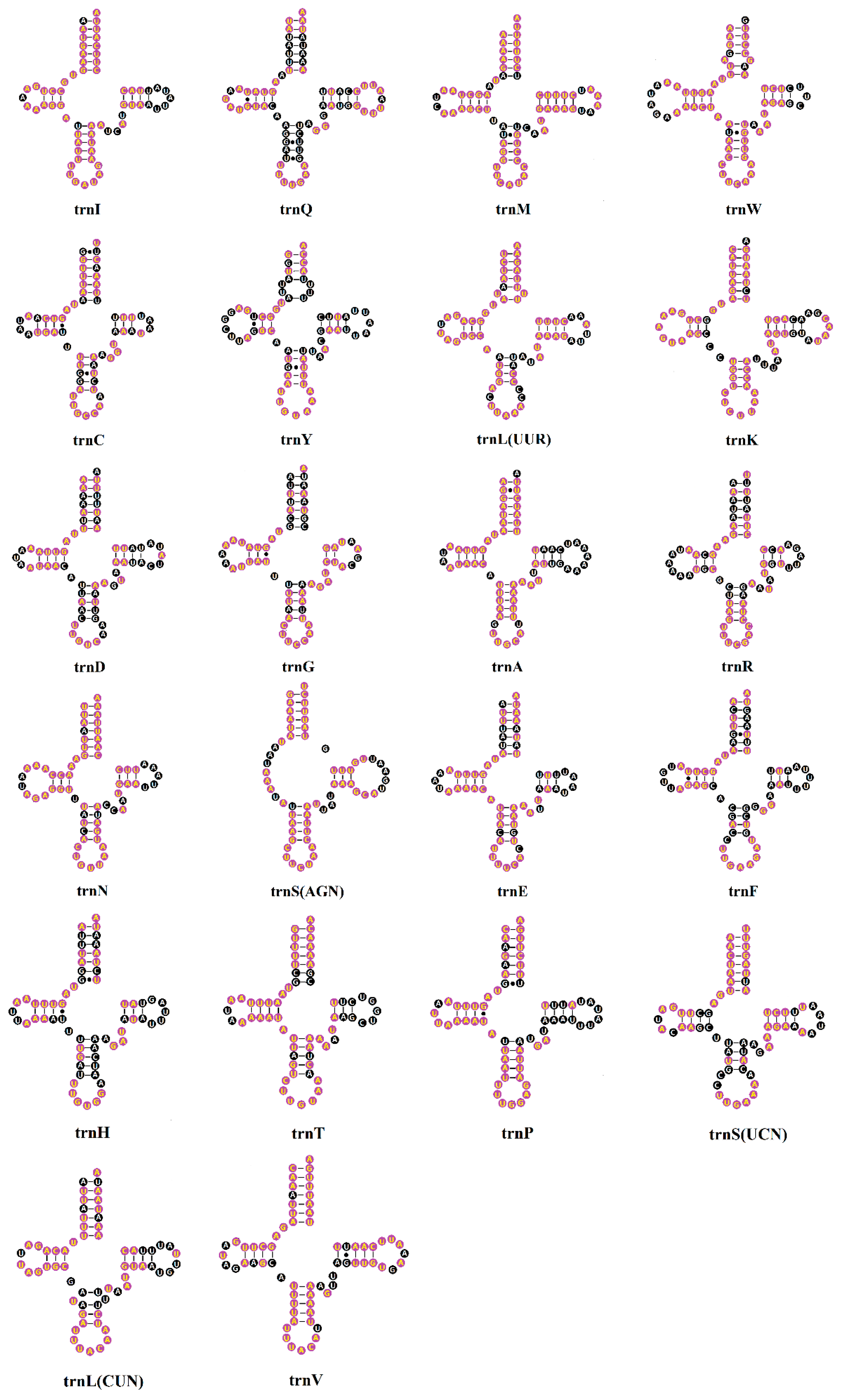
2.3. Transfer RNAs (tRNAs)
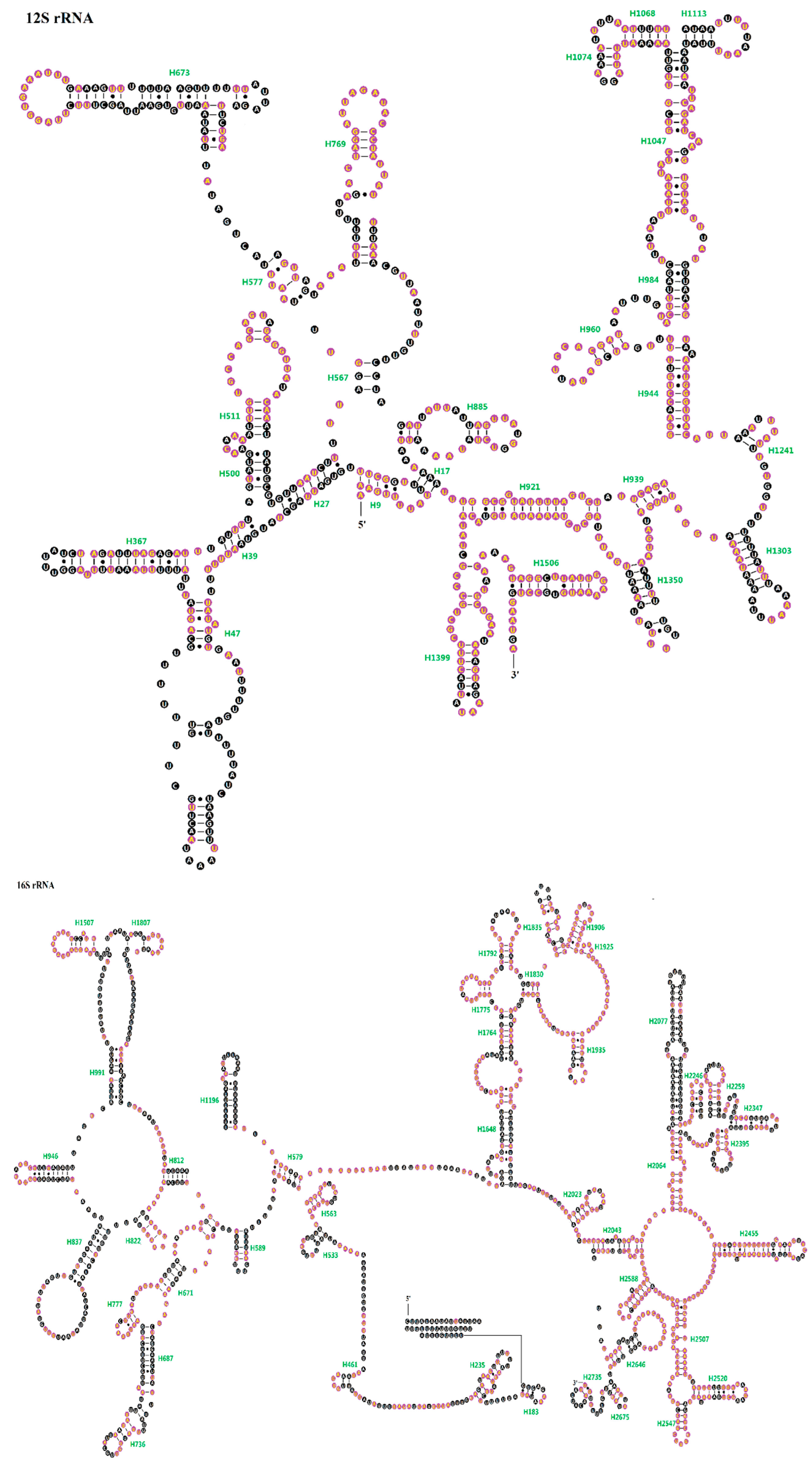
2.4. Ribosomal RNAs (rRNAs)
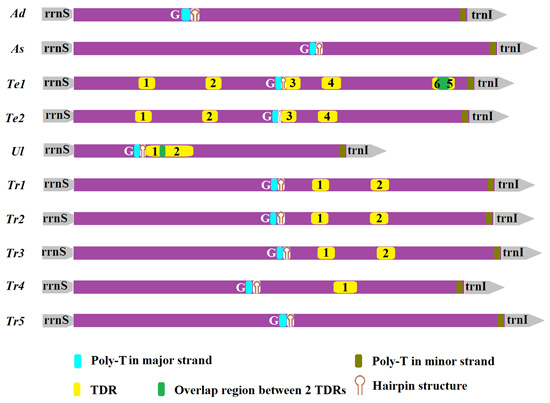
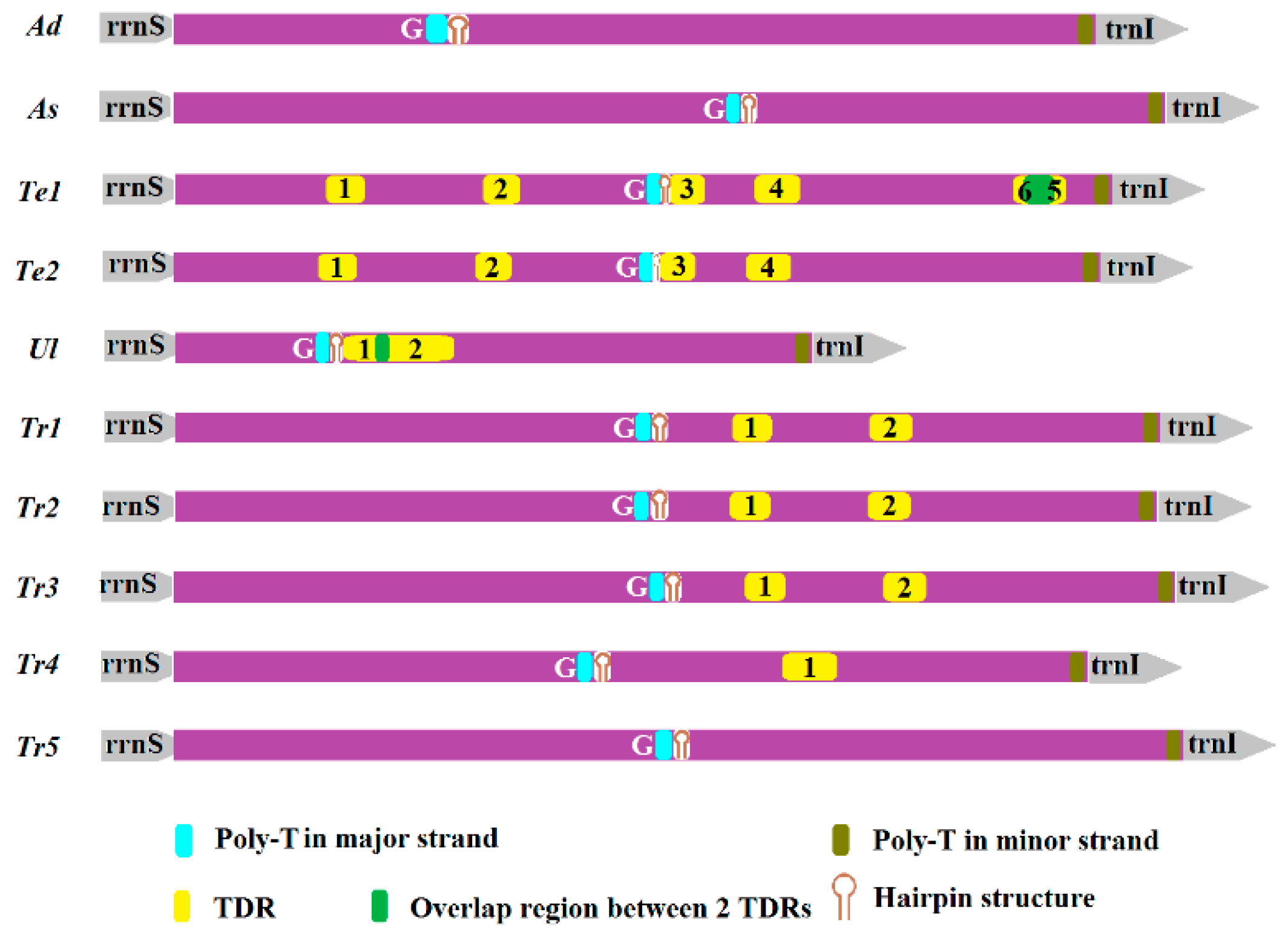
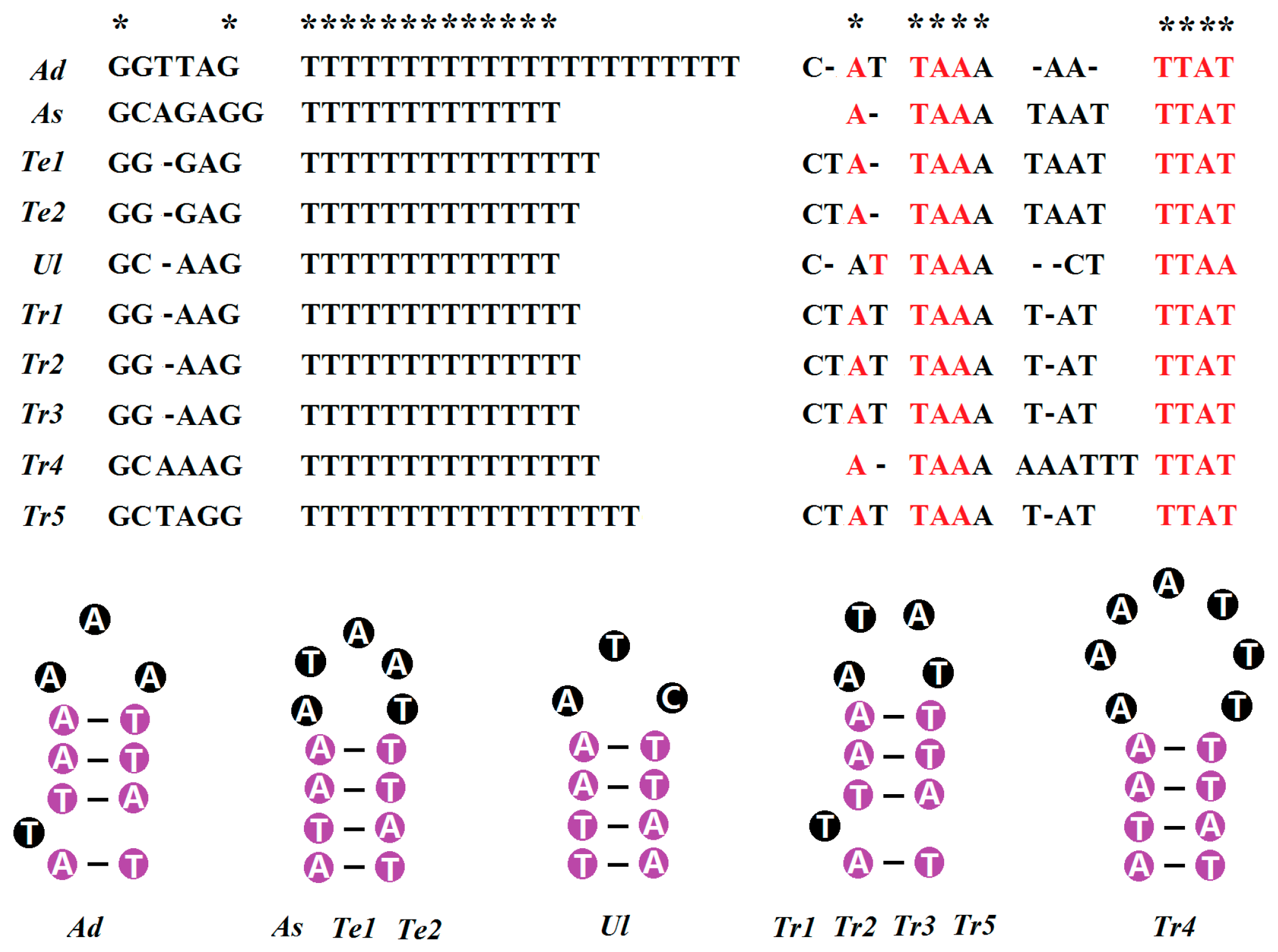
2.5. Non-Coding Regions
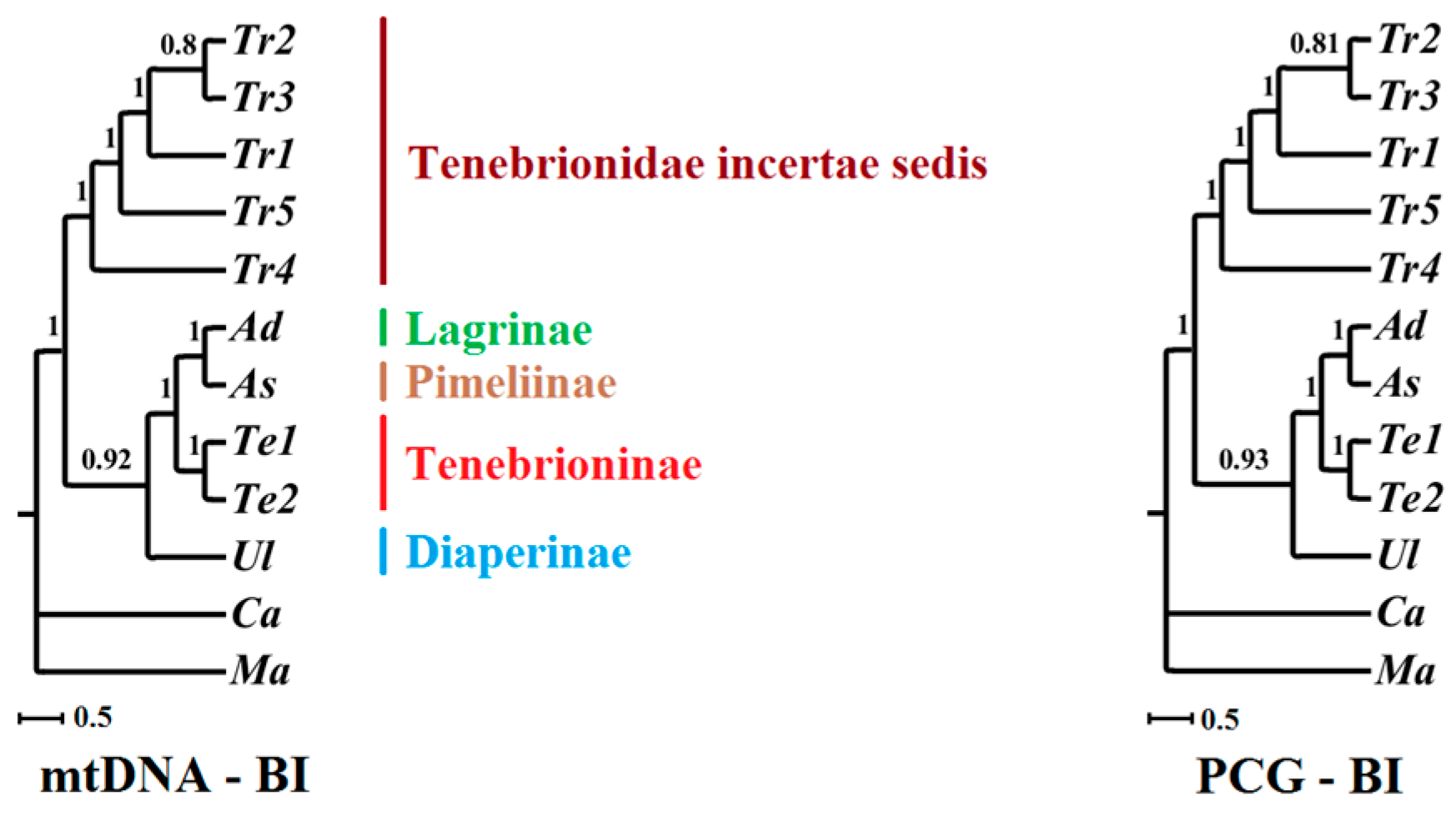
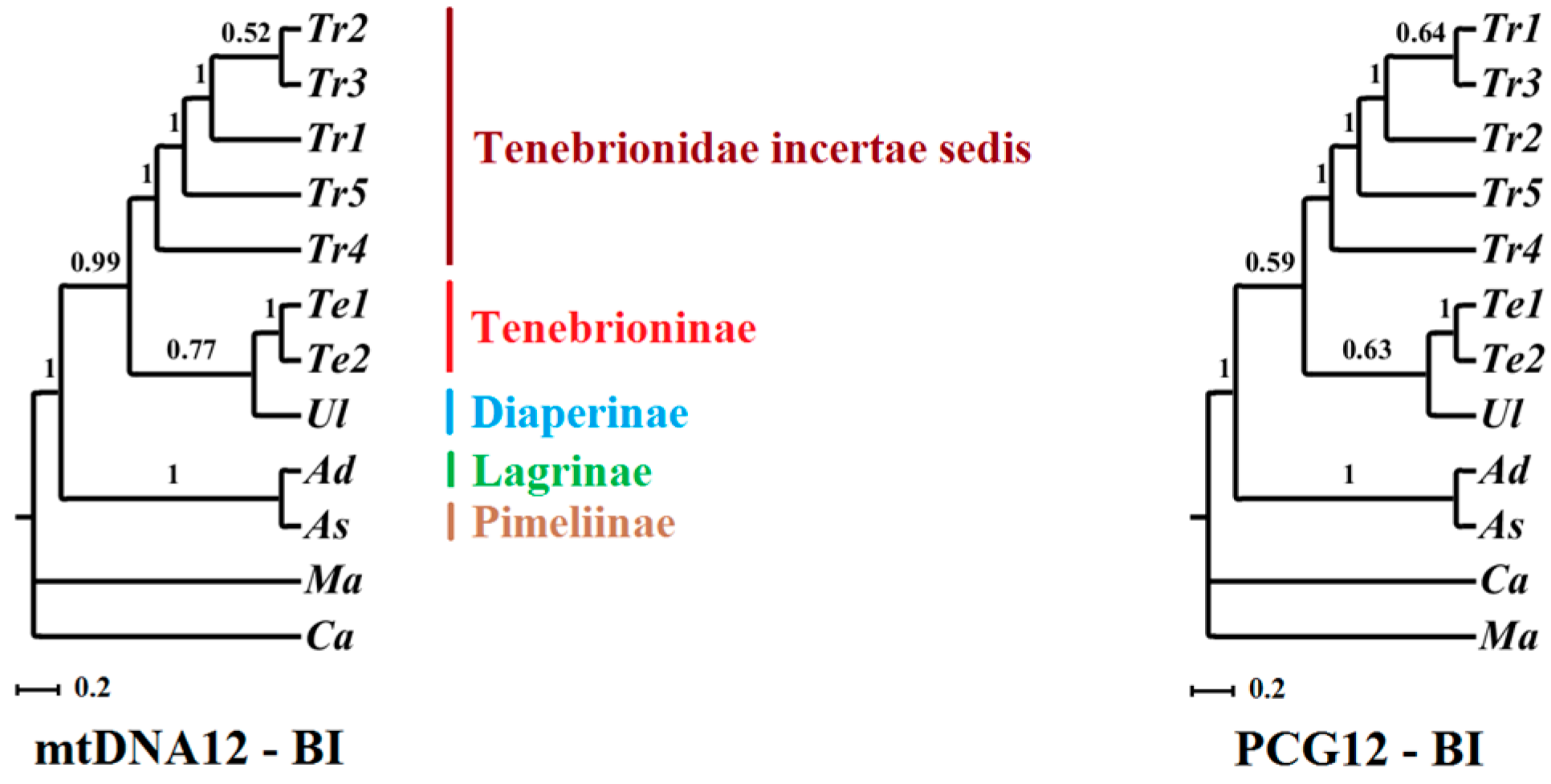
2.6. Phylogenetic Implications
3. Discussion
3.1. Mitochondrial Coding Genes
3.2. Non-Coding Regions
3.3. Proper Dataset in Phylogenetic Analyses
4. Methods
4.1. Bioinformatics Analysis
4.2. Construction of Secondary Structures of RNAs
4.3. Phylogenetic Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Boore, J.L. The use of genome-level characters for phylogenetic reconstruction. Trends Ecol. Evol. 2006, 21, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Buckley, T.R.; Frati, F.; Stewart, J.B.; Beckenbach, A.T. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 545–579. [Google Scholar] [CrossRef]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.F.; Shi, M.; Chen, X.X. Population genetic structure of Chilo suppressalis (Walker) (Lepidoptera: Crambidae): Strong subdivision in China inferred from microsatellite markers and mtDNA gene sequences. Mol. Ecol. 2008, 17, 2880–2897. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L.; Lambkin, C.L.; Barker, S.C.; Whiting, M.F. Utility of mitochondrial genomes as phylogenetic markers for insect intraordinal relationships. A case study from flies (Diptera). Syst. Entomol. 2007, 32, 40–59. [Google Scholar] [CrossRef]
- Fenn, J.D.; Song, H.; Cameron, S.L.; Whiting, M.F. A preliminary mitochondrial genome phylogeny of Orthoptera (Insecta) and approaches to maximizing phylogenetic signal found within mitochondrial genome data. Mol. Phylogenet. Evol. 2008, 49, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Dowton, M.; Cameron, S.L.; Austin, A.D.; Whiting, M.F. Phylogenetic approaches for the analysis of mitochondrial genome sequences data in the Hymenoptera—A lineage with both rapidly and slowly evolving mitochondrial genomes. Mol. Phylogenet. Evol. 2009, 52, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Leschen, R.A.B.; Beutel, R.G.; Lawrence, J.F. Coleoptera, beetles. Volume 2: Morphology and systematics (Elateroidea, Bostrichiformia, Cucujiformia partim). In Handbook of Zoology; Kristensen, N.P., Beutel, R.G., Eds.; De Gruyter: Berlin, Germany, 2010; pp. 487–491. [Google Scholar]
- Gibson, A.; Gowri-Shankar, V.G.; Higgs, P.G.; Rattray, M. A comprehensive analysis of mammalian mitochondrial genome base composition and improved phylogenetic methods. Mol. Biol. Evol. 2004, 22, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Higgs, P.G. Codon usage in mitochondrial genomes: Distinguishing context-dependent mutation from translational selection. Mol. Biol. Evol. 2007, 25, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.; Cedergren, R. Structural compensation in atypical mitochondrial tRNAs. Nat. Struct. Biol. 1994, 1, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, H.; Shi, A.M.; Štys, P.; Zhou, X.G.; Cai, W. The complete mitochondrial genome and novel gene arrangement of the unique-headed bug Stenopirates sp. (Hemiptera: Enicocephalidae). PLoS ONE 2012, 7, e29419. [Google Scholar] [CrossRef] [PubMed]
- Negrisolo, E.; Babbucci, M.; Patarnello, T. The mitochondrial genome of the ascalaphid owlfly Libelloides macaronius and comparative evolutionary mitochondriomics of neuropterid insects. BMC Genom. 2011, 12, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Rand, D.M. Endotherms, ectotherms, and mitochondrial genome-size variation. J. Mol. Evol. 1993, 37, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.L.; Wei, D.D.; Wang, B.J.; Dou, W.; Wang, J.J. The complete mitochondrial genome of the citrus red mite Panonychus citri (Acari: Tetranychidae): High genome rearrangement and extremely truncated tRNAs. BMC Genom. 2010, 11, 597. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Okada, M. Complete cDNA sequence encoding mitochondrial large ribosomal RNA of Drosophila melanogaster. Nucleic Acids Res. 1990, 18, 4592. [Google Scholar] [CrossRef] [PubMed]
- Clary, D.O.; Wolstenholme, D.R. Drosophila mitochondrial DNA: Conserved sequences in the A + T-rich region and supporting evidence for a secondary structure model of the small ribosomal RNA. J. Mol. Evol. 1987, 25, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L.; Whiting, M.F. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 2008, 408, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Gao, C.; Cui, Y.; Xie, Q.; Bu, W. The complete mitochondrial genome of the stalk-eyed bug Chauliops fallax Scott, and the monophyly of Malcidae (Hemiptera: Heteroptera). PLoS ONE 2013, 8, e55381. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.L.; Zhang, Q.L.; Guo, Z.L.; Wang, J.; Shen, Y.Y. Comparative mitogenomic analysis of the superfamily Pentatomoidea (Insecta: Hemiptera: Heteroptera) and phylogenetic implications. BMC Genom. 2015, 16, 460. [Google Scholar] [CrossRef] [PubMed]
- Page, R.D.M. Comparative analysis of secondary structure of insect mitochondrial small subunit ribosomal RNA using maximum weighted matching. Nucleic Acids Res. 2000, 28, 3839–3845. [Google Scholar] [CrossRef] [PubMed]
- Buckley, T.R.; Simon, C.; Flook, P.K.; Misof, B. Secondary structure and conserved motifs of the frequently sequenced domains IV and V of the insect mitochondrial large subunit rRNA gene. Insect. Mol. Biol. 2000, 9, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.S.; Hewitt, F.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–210. [Google Scholar] [CrossRef]
- Beutel, R.G.; Friedrich, F. Comparative study of larvae of Tenebrionoidea (Coleoptera: Cucujiformia). Eur. J. Entomol. 2005, 102, 241–264. [Google Scholar] [CrossRef]
- Hunt, T.; Bergsten, J.; Levkanicova, Z.; Papadopoulou, A.; John, O.S.; Wild, R.; Hammond, P.M.; Ahrens, D.; Balke, M.; Caterino, M.S.; et al. A comprehensive phylogeny of beetles reveals the evolutionary origins of a superradiation. Science 2007, 318, 1913–1916. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.D.; Freeland, S.J.; Landweber, L.F. A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biol. 2001, 2, 1–13. [Google Scholar]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Roberti, M.; Polosa, P.L.; Bruni, F.; Musicco, C.; Gadaleta, M.N.; Cantatore, P. DmTTF, a novel mitochondrial transcription termination factor that recognises two sequences of Drosophila melanogaster mitochondrial DNA. Nucleic Acids Res. 2003, 31, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Broughton, R.E.; Dowling, T.E. Length variation in mitochondrial DNA of the minnow Cyprinella spiloptera. Genetics 1994, 138, 179–190. [Google Scholar] [PubMed]
- Buroker, N.E.; Brown, J.R.; Gilbert, T.A.; O’Hara, P.J.; Beckenbach, A.T.; Thomas, W.K.; Smith, M.J. Length heteroplasmy of sturgeon mitochondrial DNA: An illegitimate elongation model. Genetics 1990, 124, 157–163. [Google Scholar] [PubMed]
- Savolainen, P.; Arvestad, L.; Lundeberg, J. mtDNA tandem repeats in domestic dogs and wolves: Mutation mechanism studied by analysis of the sequence of imperfect repeats. Mol. Biol. Evol. 2000, 17, 474–488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Szymura, J.M.; Hewitt, G.M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 1995, 40, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.L.; Farr, C.L.; Farquhar, A.L.; Kaguni, L.S. Sequence, organization, and evolution of the A + T region of Drosophila melanogaster mitochondrial DNA. Mol. Biol. Evol. 1994, 11, 523–538. [Google Scholar] [PubMed]
- Saito, S.; Tamura, K.; Aotsuka, T. Replication origin of mitochondrial DNA in insects. Genetics 2005, 171, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.W. Origin and evolution of mitochondrial DNA. Annu. Rev. Cell Biol. 1989, 5, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.J.; Ye, H.Y.; Huang, Y.; Shi, F.M. The phylogeny of Orthoptera inferred from mtDNA and description of Elimaea cheni (Tettigoniidae: Phaneropterinae) mitogenome. J. Genet. Genom. 2010, 37, 315–324. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Cannone, J.J.; Subramanian, S.; Schnare, M.N.; Collett, J.R.; D’Souza, L.M.; Du, Y.; Feng, B.; Lin, N.; Madabusi, L.V.; Müller, K.M.; et al. The comparative RNA web (CRW) site: An online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinform. 2002, 3, 1–31. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL X Windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- MrModeltest v2. Available online: https://github.com/nylander/MrModeltest2 (accessed on 25 May 2016).
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamily | Species | Accession Number | Abbreviation |
|---|---|---|---|
| Lagrinae | Adelium sp. | NC_013554 | Ad |
| Pimeliinae | Asbolus verrucosus | NC_027256 | As |
| Tenebrioninae | Tenebrio molitor | NC_024633 | Te1 |
| Tenebrioninae | Tenebrio molitor | KP994554 | Te2 |
| Diaperinae | Ulomoides dermestoides | NC_025332 | Ul |
| Tenebrionidae incertae sedis | Tribolium castaneum | NC_003081 | Tr1 |
| Tenebrionidae incertae sedis | Tribolium castaneum | KM009121 | Tr2 |
| Tenebrionidae incertae sedis | Tribolium castaneum | KM244661 | Tr3 |
| Tenebrionidae incertae sedis | Tribolium confusum | NC_026702 | Tr4 |
| Tenebrionidae incertae sedis | Tribolium audax | NC_024600 | Tr5 |
| Outgroup (Carabinae) | Calosoma sp. | GU176340 | Ca |
| Outgroup (Gyrinidae) | Macrogyrus oblongus | FJ859901 | Ma |
| Species | TDRs | Consensus Size (bp) | Copy Number | Position in AT-Rich Region | AT% | Percent Matches | Stem-Loop | Average ΔG | |
|---|---|---|---|---|---|---|---|---|---|
| Te1 | Te1_1 | 20 | 2.2 | 192–236 | 5′ | 85.00 | 84 | – | – |
| Te1_2 | 22 | 1.9 | 389–430 | M | 100.00 | 85 | – | – | |
| Te1_3 | 22 | 1.9 | 622–662 | M | 100.00 | 90 | 1 | 1.09 | |
| Te1_4 | 24 | 2.3 | 730–783 | M | 95.83 | 83 | 1 | −2.05 | |
| Te1_5 | 16 | 2.9 | 1070–1118 | 3′ | 93.75 | 77 | – | – | |
| Te1_6 | 24 | 2 | 1057–1103 | 3′ | 87.50 | 84 | – | – | |
| Te2 | Te2_1 | 20 | 2.2 | 183–227 | 5′ | 85.00 | 84 | – | – |
| Te2_2 | 22 | 1.9 | 380–421 | M | 100.00 | 85 | – | – | |
| Te2_3 | 22 | 1.9 | 612–652 | M | 100.00 | 90 | 1 | 1.09 | |
| Te2_4 | 24 | 2.3 | 720–773 | M | 95.83 | 83 | 1 | −2.05 | |
| Ul | Ul_1 | 14 | 3.6 | 215–267 | 5′ | 100.00 | 75 | 1 | −0.50 |
| Ul_2 | 44 | 2.1 | 255–350 | M | 97.73 | 86 | 2 | −4.40 | |
| Tr1 | Tr1_1 | 17 | 2.8 | 702–748 | M | 100.00 | 81 | – | – |
| Tr1_2 | 18 | 2.7 | 875–924 | 3′ | 100.00 | 74 | – | – | |
| Tr2 | Tr2_1 | 17 | 2.8 | 700–746 | M | 100.00 | 81 | – | – |
| Tr2_2 | 18 | 2.7 | 873–922 | 3′ | 100.00 | 74 | – | – | |
| Tr3 | Tr3_1 | 17 | 2.8 | 719–765 | M | 100.00 | 81 | – | – |
| Tr3_2 | 18 | 2.7 | 892–941 | 3′ | 100.00 | 74 | – | – | |
| Tr4 | Tr4_1 | 20 | 3.2 | 767–831 | 3′ | 95.00 | 75 | – | – |
| Species | T Stretch | Microsatellite AT | ||||||
|---|---|---|---|---|---|---|---|---|
| Position in Major Strand | Position in Minor Strand | Positions (Size) in Major Strand | ||||||
| Ad | 319–340 | 290–299 | 381–392 (6) | 433–444 (6) | ||||
| As | 696–708 | 138–147 | 887–901 | 1211–1222 | 1233–1242 | 801–816 (8) | 782–793 (6) | |
| Te1 | 595–609 | 173–192 | 633–646 (7) | 749–760 (6) | 860–869 (5) | |||
| Te2 | 586–599 | 173–192 | 623–636 (7) | 739–750 (6) | 850–859 (5) | |||
| Ul | 180–192 | 21–30 | 104–113 | 250–265 (8) | 464–475 (6) | 285–294 (5) | ||
| Tr1 | 582–595 | 794–802 | 718–727 (5) | |||||
| Tr2 | 580–593 | 793–800 | 716–725 (5) | |||||
| Tr3 | 599–612 | 793–800 | 735–744 (5) | |||||
| Tr4 | 508–522 | 51–58 | 868–875 | 770–783 (7) | 812–821 (5) | 791–800 (5) | ||
| Tr5 | 606–622 | 69–76 | 746–755 (5) | |||||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.-L.; Liu, B.-B.; Wang, X.-Y.; Han, Z.-P.; Zhang, D.-X.; Su, C.-N. Comparative Mitogenomic Analysis of Species Representing Six Subfamilies in the Family Tenebrionidae. Int. J. Mol. Sci. 2016, 17, 841. https://doi.org/10.3390/ijms17060841
Zhang H-L, Liu B-B, Wang X-Y, Han Z-P, Zhang D-X, Su C-N. Comparative Mitogenomic Analysis of Species Representing Six Subfamilies in the Family Tenebrionidae. International Journal of Molecular Sciences. 2016; 17(6):841. https://doi.org/10.3390/ijms17060841
Chicago/Turabian StyleZhang, Hong-Li, Bing-Bing Liu, Xiao-Yang Wang, Zhi-Ping Han, Dong-Xu Zhang, and Cai-Na Su. 2016. "Comparative Mitogenomic Analysis of Species Representing Six Subfamilies in the Family Tenebrionidae" International Journal of Molecular Sciences 17, no. 6: 841. https://doi.org/10.3390/ijms17060841
APA StyleZhang, H. -L., Liu, B. -B., Wang, X. -Y., Han, Z. -P., Zhang, D. -X., & Su, C. -N. (2016). Comparative Mitogenomic Analysis of Species Representing Six Subfamilies in the Family Tenebrionidae. International Journal of Molecular Sciences, 17(6), 841. https://doi.org/10.3390/ijms17060841