
Activation of DNA Damage Response Induced by the Kaposi’s Sarcoma-Associated Herpes Virus

 ,
,  ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Role of the DNA Damage Response (DDR) Pathway in KSHV Life Cycle
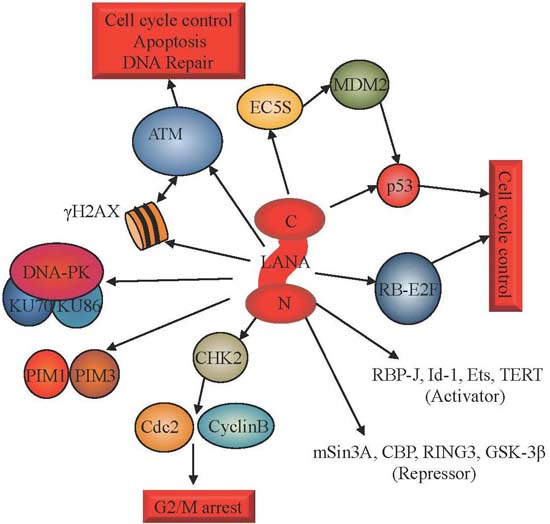
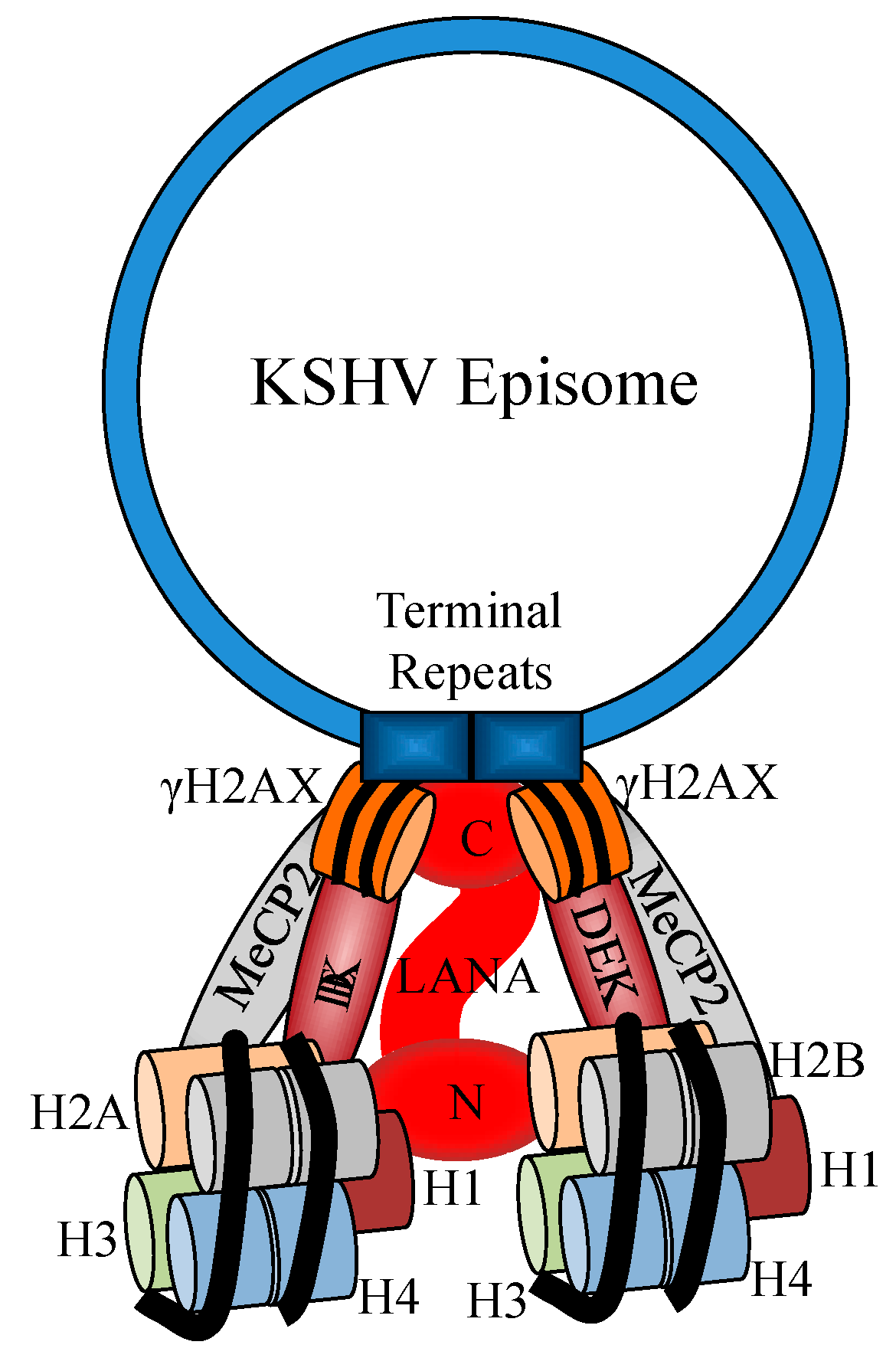
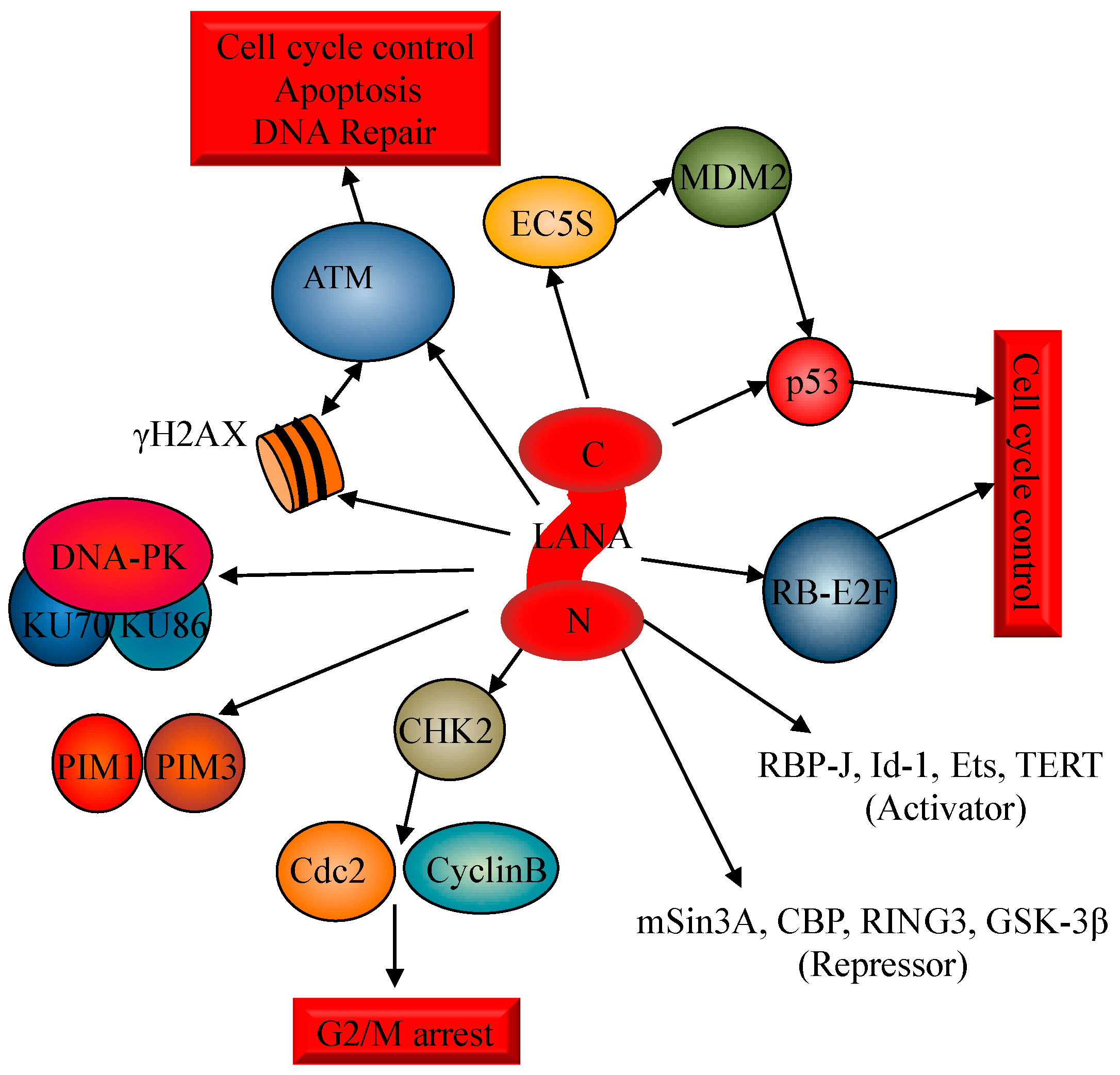
3. Activation of the DDR during KSHV Latency
4. Activation of the DDR during KSHV Lytic Replication of KSHV
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 14, 607–615. [Google Scholar] [CrossRef]
- Schottenfeld, D.; Beebe-Dimmer, J. The cancer burden attributable to biologic agents. Ann. Epidemiol. 2015, 25, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Cancian, L.; Hansen, A.; Boshoff, C. Cellular origin of Kaposi’s sarcoma and Kaposi’s sarcoma-associated herpesvirus-induced cell reprogramming. Trends Cell Biol. 2013, 23, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Rezza, G.; Tchangmena, O.B.; Andreoni, M.; Bugarini, R.; Toma, L.; Bakary, D.K.; Glikoutou, M.; Sarmati, L.; Monini, P.; Pezzotti, P.; et al. Prevalence and risk factors for human herpesvirus 8 infection in northern Cameroon. Sex Transm Dis. 2000, 27, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Serraino, D.; Toma, L.; Andreoni, M.; Buttò, S.; Tchangmena, O.; Sarmati, L.; Monini, P.; Franceschi, S.; Ensoli, B.; Rezza, G. A seroprevalence study of human herpesvirus type 8 (HHV8) in eastern and Central Africa and in the Mediterranean area. Eur. J. Epidemiol. 2001, 17, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Uldrick, T.S.; Whitby, D. Update on KSHV epidemiology, Kaposi Sarcoma pathogenesis, and treatment of Kaposi Sarcoma. Cancer Lett. 2011, 305, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, P.; Uppal, T.; Verma, S.C. Molecular biology of KSHV lytic reactivation. Viruses 2015, 7, 116–153. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.E.; Fallin, D.; Ruczinski, I.; Hutchinson, A.; Staats, B.; Vitale, F.; Lauria, C.; Serraino, D.; Rezza, G.; Mbisa, G.; et al. Associations of classic kaposi sarcoma with common variants in genes that modulate host immunity. Cancer Epidemiol. Biomark. Prev. 2006, 15, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Cavallin, L.E.; Goldschmidt-Clermont, P.; Mesri, E.A. Molecular and cellular mechanisms of KSHV oncogenesis of Kaposi’s sarcoma associated with HIV/AIDS. PLoS Pathog. 2014, 10, e1004154. [Google Scholar] [CrossRef] [PubMed]
- Rohner, E.; Wyss, N.; Heg, Z.; Faralli, Z.; Mbulaiteye, S.M.; Novak, U.; Zwahlen, M.; Egger, M.; Bohlius, J. HIV and human herpesvirus 8 co-infection across the globe: Systematic review and meta-analysis. Int. J. Cancer 2016, 138, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Boyne, J.R.; Jackson, B.R.; Taylor, A.; Macnab, S.A.; Whitehouse, A. Kaposi’s sarcoma-associated herpesvirus ORF57 protein interacts with PYM to enhance translation of viral intronless mRNAs. EMBO J. 2010, 29, 1851–1864. [Google Scholar] [CrossRef] [PubMed]
- Cordiali-Fei, P.; Latini, A.; Trento, E.; Zampatti, S.; Ferraresi, V.; Cota, C.; Volpi, S.; D’Agosto, G.; Bordignon, V.; Giardina, E.; et al. Familial Kaposi’s Sarcoma in HHV8 infected subjects presenting the G-174C allele of the IL-6 promoter: A possible role for EBV? Eur. J. Dermatol. 2014, 24, 503–504. [Google Scholar] [PubMed]
- Tornesello, M.L.; Buonaguro, L.; Cristillo, M.; Biryahwaho, B.; Downing, R.; Hatzakis, A.; Alessi, E.; Cusini, M.; Ruocco, V.; Viviano, E.; et al. MDM2 and CDKN1A gene polymorphisms and risk of kaposi’s sarcoma in african and caucasian patients. Biomarkers 2011, 16, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Thakker, S.; Verma, S.C. Co-infections and Pathogenesis of KSHV-Associated Malignancies. Front. Microbiol. 2016, 7, 151. [Google Scholar] [CrossRef] [PubMed]
- Gramolelli, S.; Schulz, T.F. The role of Kaposi sarcoma-associated herpesvirus in the pathogenesis of Kaposi sarcoma. J. Pathol. 2015, 235, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [PubMed]
- Di Domenico, E.G.; Romano, E.; Del Porto, P.; Ascenzioni, F. Multifunctional role of ATM/Tel1 kinase in genome stability: From the DNA damage response to telomere maintenance. BioMed Res. Int. 2014, 2014, 787404. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, E.; Loewer, A.; Mock, C.S.; Lahav, G. Stimulus dependent dynamics of p53 in single cells. Mol. Syst. Biol. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Murga, M.; Jaco, I.; Fan, Y.; Soria, R.; Martinez-Pastor, B.; Cuadrado, M.; Yang, S.M.; Blasco, M.A.; Skoultchi, A.I.; Fernandez-Capetillo, O. Global chromatin compaction limits the strength of the DNA damage response. J. Cell Biol. 2007, 178, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Kruhlak, M.; Crouch, E.E.; Orlov, M.; Montaño, C.; Gorski, S.A.; Nussenzweig, A.; Misteli, T.; Phair, R.D.; Casellas, R. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 2007, 447, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, L.; Rivera-Calzada, A.; Pearl, L.H.; Llorca, O. Three-dimensional structure of the humanDNA-PKcs /Ku70/Ku80 complex assembled on DNA and its implications for DNA DSB repair. Mol. Cell 2006, 22, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Usui, T.; Petrini, J.H. Taking the time to make important decisions: The checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair 2009, 8, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.; Herr, D.; Chun, J.; Xu, Y. Ser18 and 23 phosphorylation is required for p53-dependent apoptosis and tumor suppression. EMBO J. 2006, 25, 2615–2622. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B. Our cells get stressed too! Implications for human disease. Blood Cells Mol. Dis. 2007, 39, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Maya, R.; Balass, M.; Kimet, S.T.; Shkedy, D.; Leal, J.F.; Shifman, O.; Moas, M.; Buschmann, T.; Ronai, Z.; Shiloh, Y.; et al. ATM-dependent phosphorylation of Mdm2 on serine 395: Role in p53 activation by DNA damage. Genes Dev. 2001, 15, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Carson, C.T.; Schwartz, R.A.; Lilley, C.E. Interactions of viruses with the cellular DNA repair machinery. DNA Repair (Amst.) 2004, 3, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, R.; Grand, R.J. Modulation of DNA damage and repair pathways by human tumour viruses. Viruses 2015, 7, 2542–2591. [Google Scholar] [CrossRef] [PubMed]
- Boshoff, C.; Chang, Y. Kaposi’s sarcoma-associated herpesvirus: A new DNA tumor virus. Annu. Rev. Med. 2001, 52, 453–470. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Lagunoff, M.; Renne, R.; Staskus, K.; Haase, A.; Ganem, D. A cluster of latently expressed genes in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1998, 72, 8309–8315. [Google Scholar] [PubMed]
- Sun, R.; Lin, S.F.; Staskus, K.; Gradoville, L.; Grogan, E.; Haase, A.; Miller, G. Kinetics of Kaposi’s sarcoma-associated herpesvirus gene expression. J. Virol. 1999, 73, 2232–2242. [Google Scholar] [PubMed]
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposi’s sarcoma and its associated herpesvirus. Nat. Rev. Cancer. 2010, 10, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Uppal, T.; Banerjee, S.; Sun, Z.; Verma, S.C.; Robertson, E.S. KSHV lana—The master regulator of KSHV latency. Viruses 2014, 6, 4961–4998. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Garber, A.C.; Renne, R. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J. Virol. 2002, 76, 11677–11687. [Google Scholar] [CrossRef] [PubMed]
- Si, H.; Verma, S.C.; Lampson, M.A.; Cai, Q.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded LANA can interact with the nuclear mitotic apparatus protein to regulate genome maintenance and segregation. J. Virol. 2008, 82, 6734–6746. [Google Scholar] [CrossRef] [PubMed]
- Jha, H.C.; Upadhyay, S.K.; Prasaed, A.J.M.; Lu, J.; Cai, Q.; Saha, A.; Robertson, E.S. H2AX phosphorylation is important for LANA-mediated kaposi’s sarcoma-associated herpesvirus episome persistence. J. Virol. 2013, 87, 5255–5269. [Google Scholar] [CrossRef] [PubMed]
- Friborg, J., Jr.; Kong, W.; Hottiger, M.O.; Nabel, G.J. p53 Inhibition by the LANA protein of KSHV protects against cell death. Nature 1999, 402, 889–894. [Google Scholar] [PubMed]
- Radkov, S.A.; Kellam, P.; Boshoff, C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 2000, 6, 1121–1127. [Google Scholar] [PubMed]
- Si, H.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen induces chromosomal instability through inhibition of p53 function. J. Virol. 2006, 80, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Borah, S.; Robertson, E.S. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J. Virol. 2004, 78, 10348–10359. [Google Scholar] [CrossRef] [PubMed]
- Pearce, M.; Matsumura, S.; Wilson, A.C. Transcripts encoding K12, v-FLIP, v-cyclin, and the microRNA cluster of Kaposi’s sarcoma-associated herpesvirus originate from a common promoter. J. Virol. 2005, 79, 14457–14464. [Google Scholar] [CrossRef] [PubMed]
- Mittnacht, S.; Boshoff, C. Viral cyclins. Rev. Med. Virol. 2000, 10, 175–184. [Google Scholar] [CrossRef]
- Verschuren, E.W.; Jones, N.; Evan, G.I. The cell cycle and how it is steered by Kaposi’s sarcoma-associated herpesvirus cyclin. J. Gen. Virol. 2004, 85, 1347–1361. [Google Scholar] [CrossRef] [PubMed]
- Van Dross, R.; Yao, S.; Asad, S.; Westlake, G.; Mays, D.J.; Barquero, L.; Duell, S.; Pietenpol, J.A.; Browning, P.J. Constitutively active K-cyclin/CDK6 kinase in Kaposi sarcoma-associated herpesvirus-infected cells. J. Natl. Cancer Inst. 2005, 97, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Jarviluoma, A.; Ojala, P.M. Cell signaling pathways engaged by KSHV. Biochim. Biophys. Acta 2006, 1766, 140–158. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; Jung, J.U. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [PubMed]
- Guasparri, I.; Keller, S.A.; Cesarman, E. KSHV vFLIP is essential for the survival of infected lymphoma cells. J. Exp. Med. 2004, 199, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Sadler, R.; Wu, L.; Forghani, B.; Renne, R.; Zhong, W.; Herndier, B.; Ganem, D. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposiʼs sarcoma-associated herpesvirus. J. Virol. 1999, 73, 5722–5730. [Google Scholar] [PubMed]
- McCormick, C.; Ganem, D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science 2005, 307, 739–741. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E. Kaposi’s sarcoma-associated herpesvirus microRNAs. Front. Microbiol. 2012, 3, 165. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, R.P.; Sullivan, C.S. Virus-encoded microRNAs: An overview and a look to the future. PLoS Pathog. 2012, 8, e1003018. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Lu, S.; Zhang, Z.; Gonzalez, C.M.; Damania, B.; Cullen, B.R. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5570–5575. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grässer, F.A.; van Dyk, L.F.; Ho, C.K.; Shuman, S.; Chien, M.; et al. Identification of microRNAs of the herpesvirus family. Nat. Meth. 2005, 2, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Samols, M.A.; Hu, J.; Skalsky, R.L.; Renne, R. Cloning and identification of a MicroRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 9301–9305. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Lan, K.; Verma, S.C.; Si, H.; Lin, D.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus latent protein LANA interacts with HIF-1 α to upregulate RTA expression during hypoxia: Latency control under low oxygen conditions. J. Virol. 2006, 80, 7965–7975. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.; El-Guindy, A.; Countryman, J.; Ye, J.; Gradoville, L. Lytic cycle switches of oncogenic human gammaherpesviruses. Adv. Cancer Res. 2007, 97, 81–109. [Google Scholar] [PubMed]
- Ye, F.C.; Zhou, F.C.; Yoo, S.M.; Xie, J.P.; Browning, P.J.; Gao, S.J. Disruption of Kaposi’s sarcoma-associated herpesvirus latent nuclear antigen leads to abortive episome persistence. J. Virol. 2004, 78, 11121–11129. [Google Scholar] [CrossRef] [PubMed]
- Staskus, K.A.; Zhong, W.; Gebhard, K.; Herndier, B.; Wang, H.; Renne, R.; Beneke, J.; Pudney, J.; Anderson, D.J.; Ganem, D.; et al. Kaposi’s sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 1997, 71, 715–719. [Google Scholar] [PubMed]
- Jenner, R.G.; Alba, M.M.; Boshoff, C.; Kellam, P. Kaposiʼs sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 2001, 75, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, S.; Ueda, K.; Chen, J.; Okuno, T.; Yamanishi, K. Octamer-binding sequence is a key element for the autoregulation of Kaposi’s sarcoma-associated herpesvirus ORF50/Lyta gene expression. J. Virol. 2001, 75, 6894–6900. [Google Scholar] [CrossRef] [PubMed]
- Renne, R.; Zhong, W.; Herndier, B.; McGrath, M.; Abbey, N.; Kedes, D.; Ganem, D. Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 1996, 2, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Wang, H.; Herndier, B.; Ganem, D. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. USA 1996, 93, 6641–6646. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.P. Transcription profile of Kaposi’s sarcoma-associated herpesvirus in primary Kaposi’s sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 2003, 63, 2010–2015. [Google Scholar] [PubMed]
- Jeong, J.; Papin, J.; Dittmer, D. Differential regulation of the overlapping Kaposi’s sarcoma-associated herpesvirus vGCR (orf74) and LANA (orf73) promoters. J. Virol. 2001, 75, 1798–1807. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Dupin, N.; Fisher, C.; Kellam, P.; Ariad, S.; Tulliez, M.; Franck, N.; van Marck, E.; Salmon, D.; Gorin, I.; Escande, J.P.; et al. Distribution of human herpesvirus-8 latently infected cells in Kaposi’s sarcoma, multicentric Castleman’s disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. USA 1999, 96, 4546–4551. [Google Scholar] [CrossRef] [PubMed]
- Lennette, E.T.; Blackbourn, D.J.; Levy, J.A. Antibodies to human herpesvirus type 8 in the general population and in Kaposi’s sarcoma patients. Lancet 1996, 348, 858–861. [Google Scholar] [CrossRef]
- Ballestas, M.E.; Chatis, P.A.; Kaye, K.M. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 1999, 284, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Barbera, A.J.; Chodaparambil, J.V.; Kelley-Clarke, B.; Joukov, V.; Walter, J.C.; Luger, K.; Kaye, K.M. The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science 2006, 311, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Cotter, M.A.; Subramanian, C.; Robertson, E.S. The Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen binds to specific sequences at the left end of the viral genome through its carboxy-terminus. Virology 2001, 291, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Krithivas, A.; Young, D.B.; Liao, G.; Greene, D.; Hayward, S.D. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J. Virol. 2000, 74, 9637–9645. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Persson, L.M.; Wong, L.; Wilson, A.C. The latency-associated nuclear antigen interacts with MeCP2 and nucleosomes through separate domains. J. Virol. 2010, 84, 2318–2330. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Verma, S.C.; Cai, Q.; Kaul, R.; Lu, J.; Saha, A.; Robertson, E.S. Bub1 and CENP-F can contribute to Kaposi’s sarcoma-associated herpesvirus genome persistence by targeting LANA to kinetochores. J. Virol. 2010, 84, 9718–9732. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Verma, S.C.; Lu, J.; Robertson, E.S. Molecular biology of Kaposi’s sarcoma-associated herpesvirus and related oncogenesis. Adv. Virus Res. 2010, 78, 87–142. [Google Scholar] [PubMed]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Capetillo, O.; Lee, A.; Nussenzweig, M.; Nussenzweig, A. H2AX: The histone guardian of the genome. DNA Repair (Amst.) 2004, 3, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Feeney, K.M.; Parish, J.L. Targeting mitotic chromosomes: A conserved mechanism to ensure viral genome persistence. Proc. Biol. Sci. 2009, 276, 1535–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krithivas, A.; Fujimuro, M.; Weidner, M.; Young, D.B.; Hayward, S.D. Protein interactions targeting the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus to cell chromosomes. J. Virol. 2002, 76, 11596–11604. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Lan, K.; Choudhuri, T.; Robertson, E.S. Kaposi’s sarcoma associated herpesvirus-encoded latency-associated nuclear antigen modulates K1 expression through its cis-acting elements within the terminal repeats. J. Virol. 2006, 80, 3445–3458. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.V.; Dutta, D.; Ansari, M.A.; Dutta, S.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces the atm and h2ax DNA damage response early during de novo infection of primary endothelial cells, which play roles in latency establishment. J. Virol. 2014, 88, 2821–2834. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sahu, S.K.; Mohanty, S.; Chakrabarti, S.; Maji, S.; Reddy, R.R.; Jha, A.K.; Goswami, C.; Kundu, C.N.; Rajasubramaniam, S.; et al. Kaposi sarcoma herpes virus latency associated nuclear antigen protein release the g2/m cell cycle blocks by modulating atm/atr mediated checkpoint pathway. PLoS ONE 2014, 9, e100228. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Gordon, G.M.; Muller, M.G.; Dahiya, M.; Foreman, K.E. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen induces expression of the helix-loop-helix protein Id-1 in human endothelial cells. J. Virol. 2003, 77, 5975–5984. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Kuppers, D.A.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J. Virol. 2005, 79, 3468–3478. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Yamagoe, S.; Noguchi, K.; Takebe, Y.; Takahashi, N.; Uehara, Y.; Fukazawa, H. Ets-1-dependent expression of vascular endothelial growth factor receptors is activated by latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus through interaction with Daxx. J. Biol. Chem. 2006, 281, 28113–28121. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Gwack, Y.; Hwang, S.; Kim, S.; Choe, J. The transcriptional activity of cAMP response element-binding protein-binding protein is modulated by the latency associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus. J. Biol. Chem. 2001, 276, 31016–31022. [Google Scholar] [CrossRef] [PubMed]
- Fujimuro, M.; Hayward, S.D. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus manipulates the activity of glycogen synthase kinase-3β. J. Virol. 2003, 77, 8019–8030. [Google Scholar] [CrossRef] [PubMed]
- Berkovich, E.; Ginsberg, D. ATM is a target for positive regulation by E2F-1. Oncogene 2003, 22, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Sarek, G.; Kurki, S.; Enbäck, J.; Iotzova, G.; Haas, J.; Laakkonen, P.; Laiho, M.; Ojala, P.M. Reactivation of the p53 pathway as a treatment modality for KSHV-induced lymphomas. J. Clin. Investig. 2007, 117, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Fujimuro, M.; Liu, J.; Zhu, J.; Yokosawa, H.; Hayward, S.D. Regulation of the interaction between glycogen synthase kinase 3 and the Kaposi’s sarcoma-associated herpes virus latency-associated nuclear antigen. J. Virol. 2005, 79, 10429–10441. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Martin, H.; Shamay, M.; Woodard, C.; Tang, Q.Q.; Hayward, S.D. Kaposi’s sarcoma-associated herpesvirus LANA protein downregulates nuclear glycogen synthase kinase 3 activity and consequently blocks differentiation. J. Virol. 2007, 81, 4722–4731. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Weidner-Glunde, M.; Varjosalo, M.; Rainio, E.M.; Lehtonen, A.; Schulz, T.F.; Koskinen, P.J.; Taipale, J.; Ojala, P.M. KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog. 2009, 5, e1000324. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.; Lim, C.; Lee, J.Y.; Song, Y.J.; Park, J.; Choe, J.; Seo, T. DNA-PK/Ku complex binds to latency-associated nuclear antigen and negatively regulates Kaposi’s sarcoma-associated herpesvirus latent replication. Biochem. Biophys. Res. Commun. 2010, 394, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Platt, G.M.; Simpson, G.R.; Mittnacht, S.; Schulz, T.F. Latent nuclear antigen of Kaposi’s sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J. Virol. 1999, 73, 9789–9795. [Google Scholar] [PubMed]
- Nikitin, P.A.; Luftig, M.A. The DNA damage response in viral-induced cellular transformation. Br. J. Cancer 2012, 106, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Ohta, T.; Venkitaraman, A.R. A mitotic role for the DNA damage-responsive CHK2 kinase. Nat. Cell Biol. 2010, 12, 424–425. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Ertych, N.; Kienitz, A.; Vogel, C.; Schneider, V.; Fritz, B.; Jacob, R.; Dittmar, G.; Weichert, W.; Petersen, I.; Bastians, H. The CHK2-BRCA1 tumour suppressor pathway ensures chromosomal stability in human somatic cells. Nat. Cell Biol. 2010, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Koopal, S.; Furuhjelm, J.H.; Jarviluoma, A.; Jaamaa, S.; Pyakurel, P.; Pussinen, C.; Wirzenius, M.; Biberfeld, P.; Alitalo, K.; Laiho, M.; et al. Viral oncogene-induced D damage response is activated in kaposi sarcoma tumorigenesis. PLoS Pathog. 2007, 3, 1348–1360. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lee, H.; Yoon, D.W.; Albrecht, J.C.; Fleckenstein, B.; Neipel, F.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus encodes a functional cyclin. J. Virol. 1997, 71, 1984–1991. [Google Scholar] [PubMed]
- Godden-Kent, D.; Talbot, S.J.; Boshoff, C.; Chang, Y.; Moore, P.; Weiss, R.A.; Mittnacht, S. The cyclin encoded by Kaposi’s sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J. Virol. 1997, 71, 4193–4198. [Google Scholar] [PubMed]
- Jeffrey, P.D.; Tong, L.; Pavletich, N.P. Structural basis of inhibition of CDK–cyclin complexes by INK4 inhibitors. Genes Dev. 2000, 14, 3115–3125. [Google Scholar] [CrossRef] [PubMed]
- Direkze, S.; Laman, H. Regulation of growth signalling and cell cycle by Kaposi’s sarcoma associated herpesvirus genes. Int. J. Exp. Pathol. 2004, 85, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Verschuren, E.W.; Klefstrom, J.; Evan, G.I.; Jones, N. The oncogenic potential of kaposi’s sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in vivo. Cancer Cell 2002, 2, 229–241. [Google Scholar] [CrossRef]
- Leidal, A.M.; Cyr, D.P.; Hill, R.J.; Lee, P.W.; McCormick, C. Subversion of autophagy by Kaposi’s sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe 2012, 11, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Sarek, G.; Jarviluoma, A.; Moore, H.M.; Tojkander, S.; Vartia, S.; Biberfeld, P.; Laiho, M.; Ojala, P.M. Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency. PLoS Pathog. 2010, 6, e1000818. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tan, B.C.; Tseng, K.H.; Chuang, C.P.; Yeh, C.W.; Chen, K.D.; Lee, S.C.; Yung, B.Y. Nucleophosmin acts as a novel AP2α-binding transcriptional corepressor during cell differentiation. EMBO Rep. 2007, 8, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Byun, H.; Hwang, S.; Lim, C.; Choe, J. CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi’s sarcomaassociated herpesvirus open reading frame 50. J. Virol. 2001, 75, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, G.; Viiliäinen, J.; Turunen, M.; Diaz, R.; Lyly, L.; Pekkonen, P.; Rantala, J.; Ojala, K.; Sarek, G.; Teesalu, M.; et al. Oncogenic herpesvirus utilizes stress-induced cell cycle checkpoints for efficient lytic replication. PLoS Pathog. 2016, 12, e1005424. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.R.; Noerenberg, M.; Whitehouse, A. A novel mechanism inducing genome instability in Kaposi’s sarcoma-associated herpesvirus infected cells. PLoS Pathog. 2014, 10, e1004098. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, N.; Borden, K.L.B. mRNA export and cancer. Wiley Interdiscip. Rev. RNA 2011, 3, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Reed, R.; Cheng, H. TREX, SR proteins and export of mRNA. Curr. Opin. Cell Biol. 2005, 17, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Luna, R.; Rondón, A.G.; Aguilera, A. New clues to understand the role of THO and other functionally related factors in mRNP biogenesis. Biochim. Biophys. Acta 2012, 1819, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Schumann, S.; Jackson, B.R.; Baquero-Perez, B.; Whitehouse, A. Kaposi’s sarcoma-associated herpesvirus ORF57 protein: Exploiting all stages of viral mRNA processing. Viruses 2013, 5, 1901–1923. [Google Scholar] [CrossRef] [PubMed]
- Boyne, J.R.; Colgan, K.J.; Whitehouse, A. Recruitment of the complete hTREX complex is required for Kaposi’s sarcoma-associated herpesvirus intronless mRNA nuclear export and virus replication. PLoS Pathog. 2008, 4, e1000194. [Google Scholar] [CrossRef] [PubMed]
- Ganem, D. KSHV infection and the pathogenesis of Kaposi’s sarcoma. Annu. Rev. Pathol. 2006, 1, 273–296. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Damania, B. The viral interferon regulatory factors of KSHV: Immunosuppressors or oncogenes? Front. Immunol. 2011, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Damania, B.; Alvarez, X.; Ogryzko, V.; Ozato, K.; Jung, J.U. Inhibition of p300 histone acetyltransferase by viral interferon regulatory factor. Mol. Cell. Biol. 2000, 20, 8254–8263. [Google Scholar] [CrossRef] [PubMed]
- Hew, K.; Dahlroth, S.L.; Venkatachalam, R.; Nasertorabi, F.; Lim, B.T.; Cornvik, T.; Nordlund, P. The crystal structure of the DNA-binding domain of vIRF-1 from the oncogenic KSHV reveals a conserved fold for DNA binding and reinforces its role as a transcription factor. Nucleic Acids Res. 2013, 41, 4295–4306. [Google Scholar] [CrossRef] [PubMed]
- Sen, G.C.; Ransohoff, R.M. Interferon-induced antiviral actions and their regulation. Adv. Virus Res. 1992, 42, 57–102. [Google Scholar]
- Seo, Y.R.; Kelley, M.R.; Smith, M.L. Selenomethionine regulation of p53 by a ref1-dependent redox mechanism. Proc. Natl. Acad. Sci. USA 2002, 99, 14548–14553. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.C.; Nakamura, H.; Liang, X.; Feng, P.; Chang, H.; Kowalik, T.F.; Jung, J.U. Inhibition of the ATM/p53 signal transduction pathway by Kaposi’s sarcoma-associated herpesvirus interferon regulatory factor 1. J. Virol. 2006, 80, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Chavoshi, S.; Egorova, O.; Lacdao, I.K.; Farhadi, S.; Sheng, Y.; Saridakis, V. Identification of kaposi sarcoma herpesvirus (KSHV) vIRF1 protein as a novel interaction partner of human deubiquitinase USP7. J. Biol. Chem. 2016, 291, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Krenning, L.; Feringa, F.M.; Shaltiel, I.A.; van den Berg, J.; Medema, R.H. Transient activation of p53 in G2 phase is sufficient to induce senescence. Mol. Cell 2014, 55, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.R.; Chandran, B. Characterization of human herpesvirus 8 ORF59 protein (PF-8) and mapping of the processivity and viral DNA polymerase-interacting domains. J. Virol. 2000, 74, 10920–10929. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Chen, J.; Liao, Q.; Wu, Y.; Peng, C.; Chen, X. Lytic infection of Kaposi’s sarcoma-associated herpesvirus induces DNA double-strand breaks and impairs non-homologous end joining. J. Gen. Virol. 2013, 94, 1870–1875. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr. Med. Chem. Anti-Cancer Agents 2002, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Richards, K.; Damania, B. Treatment of Kaposi sarcoma associated herpesvirus-associated cancers. Front. Microbiol. 2012, 3, 141. [Google Scholar] [CrossRef] [PubMed]
- Duggan, S.T.; Keating, G.M. Pegylated liposomal doxorubicin: A review of its use in metastatic breast cancer, ovarian cancer, multiple myeloma and AIDS-related Kaposi’s sarcoma. Drugs 2011, 71, 2531–2558. [Google Scholar] [CrossRef] [PubMed]
- Gualdi, G.; Monari, P.; Fantini, F.; Cesinaro, A.M.; Cimitan, A. Elettrochemotherapy-induced virus disappearance in HHV-8 positive skin nodules of Kaposi’s sarcoma: First histological and immunohistochemical demonstration of efficacy. J. Eur. Acad. Dermatol. Venereol. 2010, 24, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Latini, A.; Bonadies, A.; Trento, E.; Bultrini, S.; Cota, C.; Solivetti, F.M.; Ferraro, C.; Ardigò, M.; Amorosi, B.; Palamara, G.; et al. Effective treatment of Kaposi’s sarcoma by electrochemotherapy and intravenous bleomycin administration. Dermatol. Ther. 2012, 25, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Lattif, A.A.; Xie, J.; Weinberg, A.; Gao, S. Nutlin-3 induces apoptosis, disrupts viral latency and inhibits expression of angiopoietin-2 in Kaposi sarcoma tumor cells. Cell Cycle 2012, 11, 1393–1399. [Google Scholar] [CrossRef] [PubMed]
- Santag, S.; Jager, W.; Karsten, C.B.; Kati, S.; Pietrek, M.; Steinemann, D.; Sarek, G.; Ojala, P.M.; Schulz, T.F. Recruitment of the tumour suppressor protein p73 by Kaposi’s Sarcoma Herpes virus latent nuclear antigen contributes to the survival of primary effusion lymphoma cells. Oncogene 2013, 32, 3676–3685. [Google Scholar] [CrossRef] [PubMed]
- Curreli, F.; Friedman-Kien, A.E.; Flore, O. Glycyrrhizic acid alters Kaposi sarcoma-associated herpesvirus latency, triggering p53-mediated apoptosis in transformed B lymphocytes. J. Clin. Investig. 2005, 115, 642–652. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Domenico, E.G.; Toma, L.; Bordignon, V.; Trento, E.; D’Agosto, G.; Cordiali-Fei, P.; Ensoli, F. Activation of DNA Damage Response Induced by the Kaposi’s Sarcoma-Associated Herpes Virus. Int. J. Mol. Sci. 2016, 17, 854. https://doi.org/10.3390/ijms17060854
Di Domenico EG, Toma L, Bordignon V, Trento E, D’Agosto G, Cordiali-Fei P, Ensoli F. Activation of DNA Damage Response Induced by the Kaposi’s Sarcoma-Associated Herpes Virus. International Journal of Molecular Sciences. 2016; 17(6):854. https://doi.org/10.3390/ijms17060854
Chicago/Turabian StyleDi Domenico, Enea Gino, Luigi Toma, Valentina Bordignon, Elisabetta Trento, Giovanna D’Agosto, Paola Cordiali-Fei, and Fabrizio Ensoli. 2016. "Activation of DNA Damage Response Induced by the Kaposi’s Sarcoma-Associated Herpes Virus" International Journal of Molecular Sciences 17, no. 6: 854. https://doi.org/10.3390/ijms17060854
APA StyleDi Domenico, E. G., Toma, L., Bordignon, V., Trento, E., D’Agosto, G., Cordiali-Fei, P., & Ensoli, F. (2016). Activation of DNA Damage Response Induced by the Kaposi’s Sarcoma-Associated Herpes Virus. International Journal of Molecular Sciences, 17(6), 854. https://doi.org/10.3390/ijms17060854