
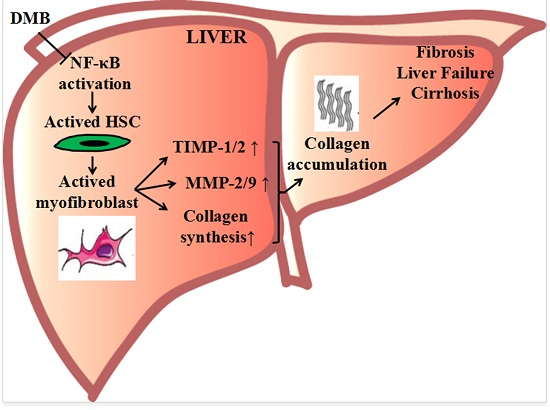
Demethyleneberberine Protects against Hepatic Fibrosis in Mice by Modulating NF-κB Signaling

Abstract
:
1. Introduction
2. Results
2.1. Demethyleneberberine (DMB) Alleviates Thioacetamide (TAA)-Induced Acute Hepatic Injury
2.2. DMB Attenuates TAA-Induced Hepatic Fibrosis
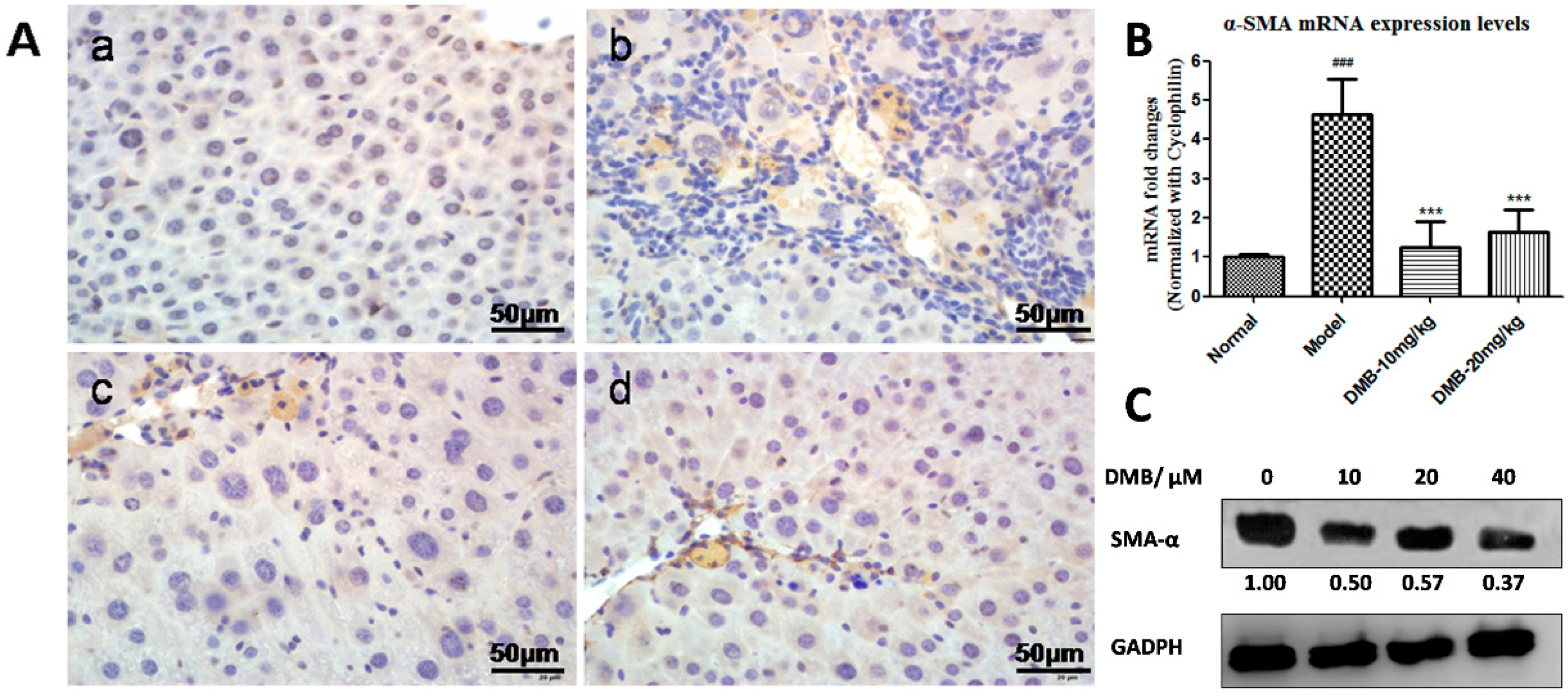
2.3. DMB Inhibits the Expression of Alpha-Smooth Muscle Actin (α-SMA)
2.4. DMB Blocks the Growth Factor β 1 (TGF-β1)-Smad Signaling
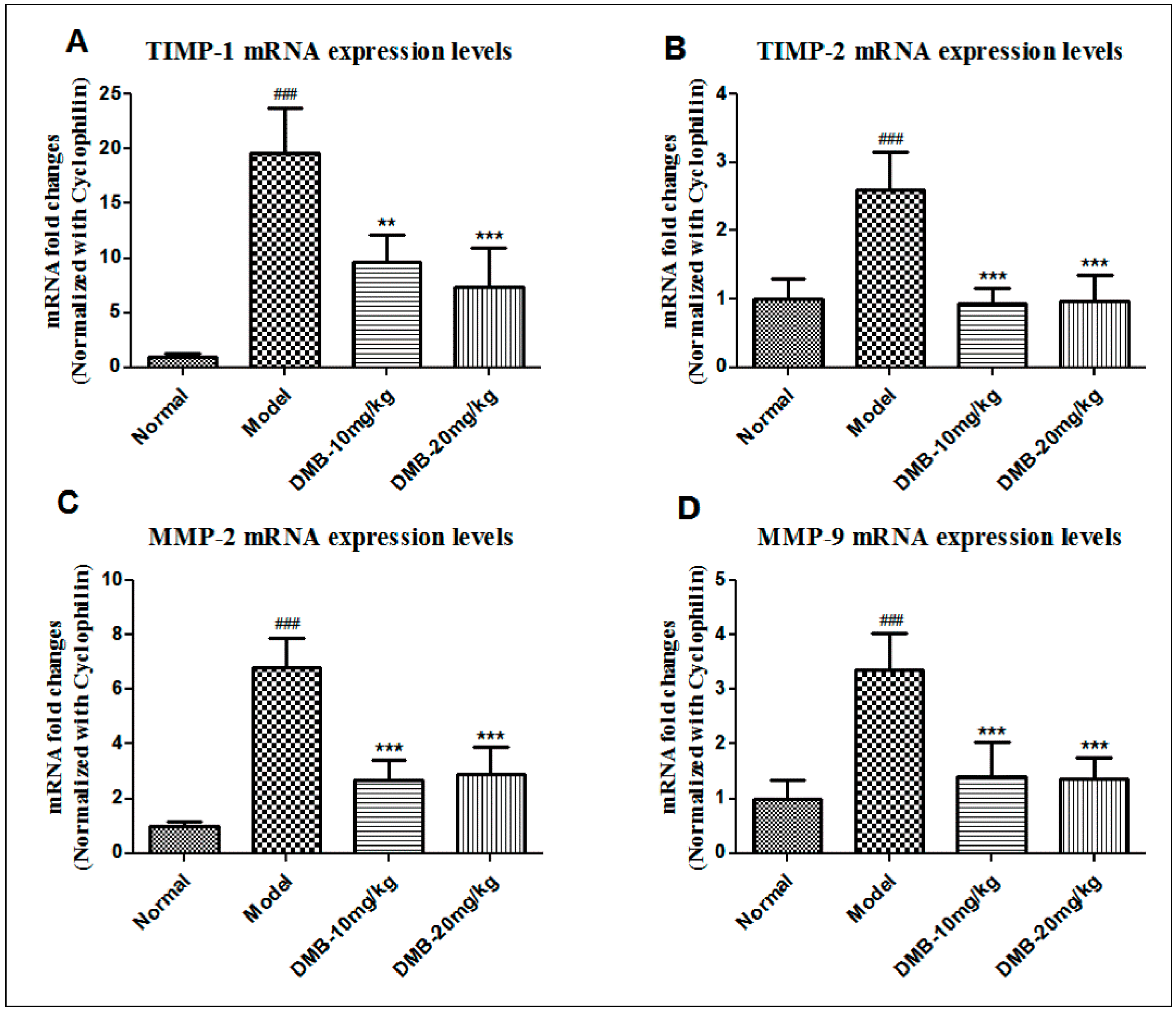
2.5. DMB Reduces the Expression of Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of MMP (TIMPs)
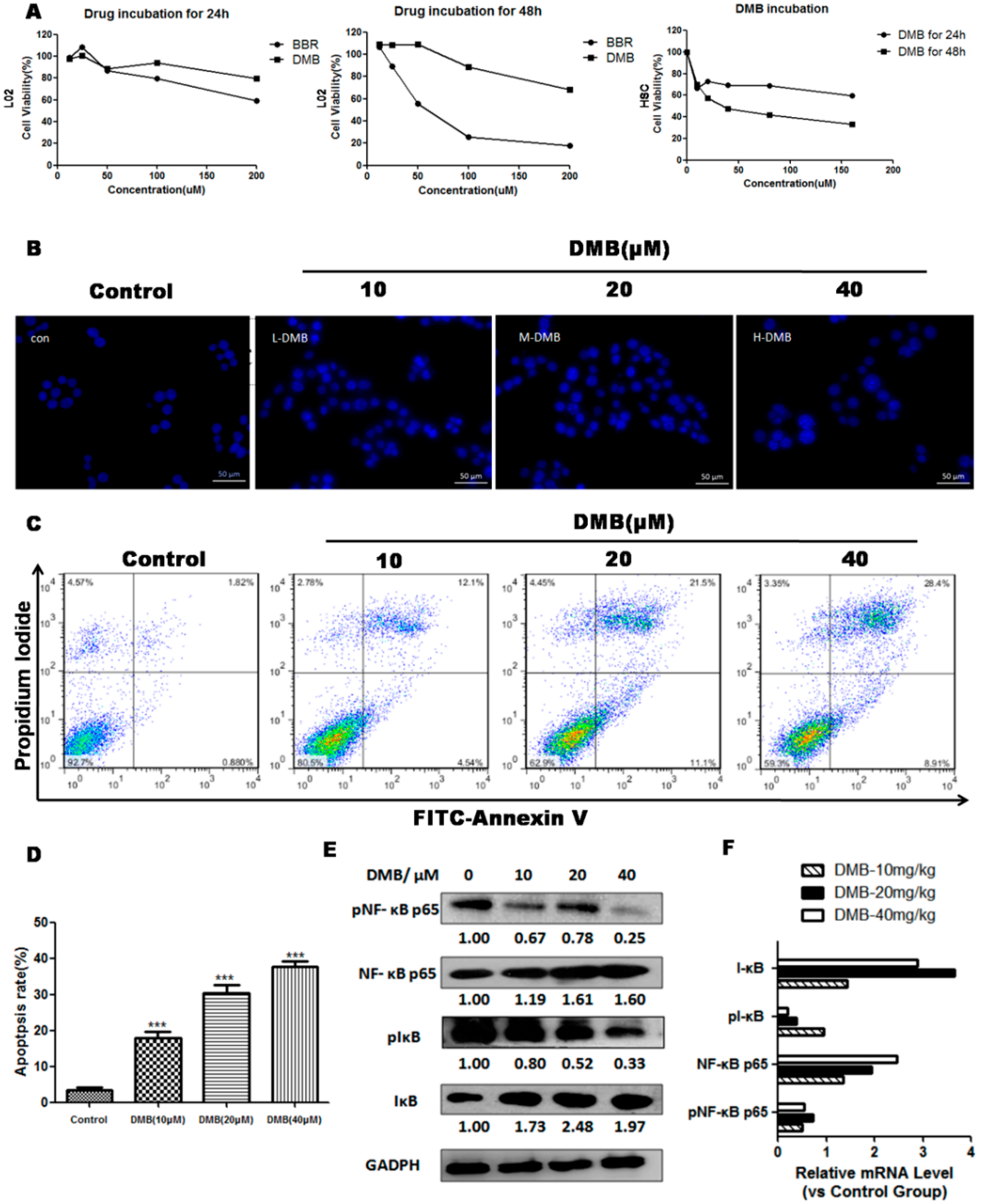
2.6. DMB Promotes Apoptosis of Hepatic Stellate Cells (HSCs) via Suppressing Active Nuclear Factor-κB (NF-κB) Activation
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Treatments
4.3. Animal Experiments
4.4. Pathological Evaluation
4.5. Biochemical Assay
4.6. Immunohistochemical Analysis
4.7. qPCR and Western Blot Analysis
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wu, X.; Zhang, F.; Xiong, X.; Lu, C.; Lian, N.; Lu, Y.; Zheng, S. Tetramethylpyrazine reduces inflammation in liver fibrosis and inhibits inflammatory cytokine expression in hepatic stellate cells by modulating NLRP3 inflammasome pathway. IUBMB Life 2015, 67, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Hautekeete, M.L.; Geerts, A. The hepatic stellate (Ito) cell: Its role in human liver disease. Virchows Arch. 1997, 430, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Friedman, S.L. Liver fibrogenesis and the role of hepatic stellate cells: New insights and prospects for therapy. J. Gastroen. Hepatol. 1999, 14, 618–633. [Google Scholar] [CrossRef]
- Suhaimi, N.A.M.; Zhuo, L. Imidazolium salt attenuates thioacetamide-induced liver fibrosis in mice by modulating inflammation and oxidative stress. Dig. Liver Dis. 2012, 44, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Di Lullo, G.A.; Sweeney, S.M.; Korkko, J.; Ala-Kokko, L.; San Antonio, J.D. Mapping the ligand-binding sites and disease-associated mutations on the most abundant protein in the human, type I collagen. J. Biol. Chem. 2002, 277, 4223–4231. [Google Scholar] [CrossRef] [PubMed]
- Kurikawa, N.; Suga, M.; Kuroda, S.; Yamada, K.; Ishikawa, H. An angiotensin II type 1 receptor antagonist, olmesartan medoxomil, improves experimental liver fibrosis by suppression of proliferation and collagen synthesis in activated hepatic stellate cells. Br. J. Pharmacol. 2003, 139, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Hemmann, S.; Graf, J.; Roderfeld, M.; Roeb, E. Expression of MMPs and TIMPs in liver fibrosis—A systematic review with special emphasis on anti-fibrotic strategies. J. Hepatol. 2007, 46, 955–975. [Google Scholar] [CrossRef] [PubMed]
- Stravitz, R.T. Critical management decisions in patients with acute liver failure. Chest 2008, 134, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.G.; Chen, A.P. Activation of peroxisome proliferator-activated receptor-γ by curcumin blocks the signaling pathways for PDGF and EGF in hepatic stellate cells. Lab. Investig. 2008, 88, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, J.; Li, S.; He, B.; Mi, Y.L.; Cao, H.C.; Zhang, C.Q.; Li, L.J. Ameliorative effect of grape seed proanthocyanidin extract on thioacetamide-induced mouse hepatic fibrosis. Toxicol. Lett. 2012, 213, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, V.; Lee, J.; Pappa, A.; Petersen, D.R. Involvement of p65 in the regulation of NF-κB in rat hepatic stellate cells during cirrhosis. Biochem. Biophys. Res. Commun. 2000, 273, 546–550. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Pu, G.; Tang, R.; Zhang, D.; Pan, W. Activation of nuclear factor κB in the hepatic stellate cells of mice with schistosomiasis japonica. PLoS ONE 2014, 9, e104323. [Google Scholar]
- Barnes, P.J.; Larin, M. Mechanisms of disease—Nuclear factor-κB—A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [PubMed]
- Benfield, C.T.O.; Mansur, D.S.; McCoy, L.E.; Ferguson, B.J.; Bahar, M.W.; Oldring, A.P.; Grimes, J.M.; Stuart, D.I.; Graham, S.C.; Smith, G.L. Mapping the I κB kinase β (IKK β)-binding interface of the B14 protein, a vaccinia virus inhibitor of IKK β-mediated activation of nuclear factor κB. J. Biol. Chem. 2011, 286, 20727–20735. [Google Scholar] [CrossRef] [PubMed]
- Rousar, T.; Kucera, O.; Krivakova, P.; Lotkova, H.; Kandar, R.; Muzakova, V.; Cervinkova, Z. Evaluation of oxidative status in acetaminophen-treated rat hepatocytes in culture. Physiol. Res. 2009, 58, 239–246. [Google Scholar] [PubMed]
- Yamamoto, K.; Sasakawa, Y.; Nakaoka, F.; Nakao, M.; Nakamura, M.; Kominami, A.; Abe, M.; Fukuhama, C.; Kagawa, K. Effect of globin digest on the liver injury and hepatic gene expression profile in galactosamine-induced liver injury in SD rats. Life Sci. 2011, 88, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.L.; Wang, Y.J.; Zhang, Q.Y.; Liu, B.; Wang, F.Y.; Li, J.J.; Zhu, R.Z. Hepatoprotective effects of baicalein against CCl4-induced acute liver injury in mice. World J. Gastroenterol. 2012, 18, 6605–6613. [Google Scholar] [CrossRef] [PubMed]
- Fitzhugh, O.G.; Nelson, A.A. Liver tumours in rats fed thiourea or thioacetamide. Science 1948, 108, 626–628. [Google Scholar] [CrossRef] [PubMed]
- Chilakapati, J.; Shankar, K.; Korrapati, M.C.; Hill, R.A.; Mehendale, H.M. Saturation toxicokinetics of thioacetamide: Role in initiation of liver injury. Drug Metab. Dispos. 2005, 33, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Wanibuchi, H.; Morimura, K.; Wongpoomchai, R.; Chusiri, Y.; Gonzalez, F.J.; Fukushima, S. Role of CYP2E1 in thioacetamide-induced mouse hepatotoxicity. Toxicol. Appl. Pharm. 2008, 228, 295–300. [Google Scholar] [CrossRef] [PubMed]
- DiezFernandez, C.; Sanz, N.; Cascales, M. Intracellular calcium concentration impairment in hepatocytes from thioacetamide-treated rats. Implications for the activity of Ca2+-dependent enzymes. J. Hepatol. 1996, 24, 460–467. [Google Scholar] [CrossRef]
- Stankova, P.; Kucera, O.; Lotkova, H.; Rousar, T.; Endlicher, R.; Cervinkova, Z. The toxic effect of thioacetamide on rat liver in vitro. Toxicol. In Vitro 2010, 24, 2097–2103. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, H.; Wada, A.; Takayanagi, H. Histological studies of liver cirrhosis in white rats by thioacetamide feeding. Gan 1956, 47, 805–807. [Google Scholar] [PubMed]
- Palacios, R.S.; Roderfeld, M.; Hemmann, S.; Rath, T.; Atanasova, S.; Tschuschner, A.; Gressner, O.A.; Weiskirchen, R.; Graf, J.; Roeb, E. Activation of hepatic stellate cells is associated with cytokine expression in thioacetamide-induced hepatic fibrosis in mice. Lab. Investig. 2008, 88, 1192–1203. [Google Scholar] [CrossRef] [PubMed]
- De David, C.; Rodrigues, G.; Bona, S.; Meurer, L.; Gonzalez-Gallego, J.; Tunon, M.J.; Marroni, N.P. Role of quercetin in preventing thioacetamide-induced liver injury in rats. Toxicol. Pathol. 2011, 39, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Shaker, M.E.; Shiha, G.E.; Ibrahim, T.M. Comparison of early treatment with low doses of nilotinib, imatinib and a clinically relevant dose of silymarin in thioacetamide-induced liver fibrosis. Eur. J. Pharmacol. 2011, 670, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Liu, P.Q.; Wu, X.Q.; Liu, W.H.; Shen, X.Y.; Lan, T.A.; Xu, S.W.; Peng, J.; Xie, X.; Huang, H.Q. Berberine attenuates lipopolysaccharide-induced extracelluar matrix accumulation and inflammation in rat mesangial cells: Involvement of NF-κB signaling pathway. Mol. Cell. Endocrinol. 2011, 331, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, B.; Liu, X.C.; Fu, X.Q.; Xiong, Z.G.; Chen, L.; Sartor, O.; Dong, Y.; Zhang, H.T. Berberine suppresses androgen receptor signaling in prostate cancer. Mol. Cancer Ther. 2011, 10, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Yang, Q.J.; Mu, Y.Q.; Zhou, L.Y.; Liu, Y.Z.; Zhou, Q.X.; He, B.C. Berberine inhibits the proliferation of colon cancer cells by inactivating Wnt/β-catenin signaling. Int. J. Oncol. 2012, 41, 292–298. [Google Scholar] [PubMed]
- Sun, X.; Zhang, X.D.; Hu, H.; Lu, Y.N.; Chen, J.; Yasuda, K.; Wang, H.Y. Berberine inhibits hepatic stellate cell proliferation and prevents experimental liver fibrosis. Biol. Pharm. Bull. 2009, 32, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Qiang, X.; Zhang, M.; Ma, D.; Zhao, Z.; Zhou, C.; Liu, X.; Li, R.; Chen, H.; Zhang, Y. Demethyleneberberine, a natural mitochondria-targeted antioxidant, inhibits mitochondrial dysfunction, oxidative stress, and steatosis in alcoholic liver disease mouse model. J. Pharmacol. Exp. Ther. 2015, 352, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, Y.; Okazaki, I. Emerging insights into transforming growth factor β Smad signal in hepatic fibrogenesis. Gut 2007, 56, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pan, Y.; Kan, M.J.; Xiao, X.N.; Wang, Y.J.; Guan, F.Y.; Zhang, X.W.; Chen, L. Hepatoprotective effects of berberine on liver fibrosis via activation of AMP-activated protein kinase. Life Sci. 2014, 98, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Zuo, F.; Nakamura, N.; Akao, T.; Hattori, M. Pharmacokinetics of berberine and its main metabolites in conventional and pseudo germ-free rats determined by liquid chromatography/ion trap mass spectrometry. Drug Metab. Dispos. 2006, 34, 2064–2072. [Google Scholar] [CrossRef] [PubMed]
- Kheir, M.M.; Wang, Y.G.; Hua, L.; Hu, J.; Li, L.L.; Lei, F.; Dua, L.J. Acute toxicity of berberine and its correlation with the blood concentration in mice. Food Chem. Toxicol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Bruck, R.; Aeed, H.; Shirin, H.; Matas, Z.; Zaide, L.; Avni, Y.; Halpern, Z. The hydroxyl radical scavengers dimethylstioxide and dimethylthiourea protect rats against thioacetamide-induced f-ant hepatic failure. J. Heputol. 1999, 31, 27–38. [Google Scholar] [CrossRef]
- Kim, K.H.; Bae, J.-H.; Cha, S.-W.; Han, S.-S.; Park, K.H.; Tae Cheon, J. Role of metabolic activation by cytochrome P450 in thioacetamide-induced suppression of antibody response in male BALB/c mice. Toxicol. Lett. 2000, 114, 225–235. [Google Scholar] [CrossRef]
- Buko, V.; Belonovskaya, E.; Naruta, E.; Lukivskaya, O.; Kanyuka, O.; Zhuk, O.; Kranc, R.; Stoika, R.; Sybirna, N. Pituitary tumor transforming gene as a novel regulatory factor of liver fibrosis. Life Sci. 2015, 132, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, X.; Zhou, Q.; Huang, C.; Meng, X.; Xu, F.; Li, J. Silent information regulator 1 (SIRT1) ameliorates liver fibrosis via promoting activated stellate cell apoptosis and reversion. Toxicol. Appl. Pharmacol. 2015, 289, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.J.; Mu, D.; Jiang, M.D.; Zheng, S.M.; Zhang, Y.; He, S.; Weng, M.; Zeng, W.Z. Hepatoprotective effect of juglone on dimethylnitrosamine-induced liver fibrosis and its effect on hepatic antioxidant defence and the expression levels of α-SMA and collagen III. Mol. Med. Rep. 2015, 12, 4095–4102. [Google Scholar] [PubMed]
- Ding, H.; Shi, J.H.; Wang, Y.; Guo, J.; Zhao, J.H.; Dong, L. Neferine inhibits cultured hepatic stellate cell activation and facilitates apoptosis A possible molecular mechanism. Eur. J. Pharmacol. 2011, 650, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.S.; Zhang, F.; Wei, D.H.; Zhu, X.J.; Zhang, X.P.; Chen, L.; Lu, Y.; Zheng, S.Z. Paeonol inhibits hepatic fibrogenesis via disrupting nuclear factor-B pathway in activated stellate cells:in vivo and in vitro studies. J. Gastroen. Hepatol. 2013, 28, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, Y.H.; Li, M.Q.; Xu, T.J.; Wang, X.L.; Hong, B.; Niu, Y.C. Curcumol induces HSC-T6 cell death through suppression of Bcl-2: Involvement of PI3K and NF-κB pathways. Eur. J. Pharm. Sci. 2014, 65, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.; Schoonhoven, R.; Tuvia, S.; Brenner, D.A.; Rippe, R.A. Nuclear factor κB in proliferation, activation, and apoptosis in rat hepatic stellate cells. J. Hepatol. 2000, 33, 49–58. [Google Scholar] [CrossRef]
- Kanzler, S.; Lohse, A.W.; Keil, A.; Henninger, J.; Dienes, H.P.; Schirmacher, P.; Rose-John, S.; Zum Buschenfelde, K.H.M.; Blessing, M. TGF-β 1 in liver fibrosis: An inducible transgenic mouse model to study liver fibrogenesis. Am. J. Physiol.-Gastrointest. Liver Physiol. 1999, 276, G1059–G1068. [Google Scholar]
- Qiang, X.; Lulu, X.; Zhang, M.; Zhang, P.; Wang, Y.; Wang, Y.; Zhao, Z.; Chen, H.; Liu, X.; Zhang, Y. Demethyleneberberine attenuates non-alcoholic fatty liver disease with activation of AMPK and inhibition of oxidative stress. Biochem. Biophys. Res. Commun. 2016, 472, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Nanji, A.A.; Mendenhall, C.; French, S.W. Beef fat prevents alcoholic liver disease in the rat. Alcohol. Clin. Exp. Res. 1989, 13, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Ruwart, M.J.; Wilkinson, K.E.; Rush, B.D.; Vidmar, T.J.; Peters, K.M.; Henley, K.S. The integrated value of serum procollagen III peptide over time predicts hepatic hydroxyproline content and stainable collagen in a model of dietary cirrhosis in the rat. Hepatology 1989, 10, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Liver Pathological Evaluation a | Average b | ||||
|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | ||
| Normal | 6 | 0 | 0 | 0 | 0 | 0 |
| TAA | 0 | 0 | 1 | 2 | 3 | 3.3 ± 0.5 ### |
| TAA + BBR (10 mg/kg) | 0 | 0 | 6 | 0 | 0 | 2.0 ± 0.0 ** |
| TAA + DMB (10 mg/kg) | 0 | 3 | 3 | 0 | 0 | 1.5 ± 0.5 *** |
| Group | ALT (IU/L) | AST (IU/L) |
|---|---|---|
| Normal | 18.0 ± 2.1 | 41.8 ± 1.8 |
| TAA | 3063.9 ± 161.6 ### | 208.0 ± 20.6 ### |
| TAA + BBR (10 mg/kg) | 1943.6 ± 372.3 *** | 123.8 ± 21.2 *** |
| TAA + DMB (10 mg/kg) | 1661.8 ± 78.3 *** | 121.9 ± 10.4 *** |
| Group | Severity Score of Liver Fibrosis a | Average b | ||||
|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | ||
| Normal | 8 | 0 | 0 | 0 | 0 | 0 |
| TAA | 0 | 0 | 0 | 2 | 6 | 3.8 ± 0.5 ### |
| DMB-10 mg/kg | 0 | 4 | 4 | 0 | 0 | 1.5 ± 0.5 *** |
| DMB-20 mg/kg | 0 | 5 | 3 | 0 | 0 | 1.4 ± 0.5 *** |
| Group | Hypdroxyproline (µg/g Wet Weight) | ALT (IU/L) | AST (IU/L) | ALB (g/L) | Body Weight (g) | Liver Weight (g) | Liver/Body Weight Ratio (%) |
|---|---|---|---|---|---|---|---|
| Normal | 5.2 ± 13.8 | 21.4 ± 5.7 | 29.9 ± 3.9 | 42.9 ± 3.0 | 39.9 ± 3.5 | 1.6 ± 0.2 | 4.4 ± 0.4 |
| TAA | 225.9 ± 26.6 ### | 151.6 ± 14.8 ### | 69.3 ± 7.7 ### | 34.6 ± 1.6 ### | 34.7 ± 3.1 ## | 2.3 ± 0.3 ### | 6.8 ± 0.5 ### |
| DMB-10 mg/kg | 119.4 ± 20.6 *** | 78.3 ± 7.3 *** | 36.3 ± 7.4 *** | 38.2 ± 1.7 * | 36.5 ± 2.2 | 1.8 ± 0.3 ** | 5.3 ± 0.3 *** |
| DMB-20 mg/kg | 117.3 ± 11.8 *** | 76.9 ± 9.9 *** | 42.5 ± 6.4 *** | 42.4 ± 1.7 *** | 36.4 ± 2.3 | 1.7 ± 0.1 *** | 5.2 ± 0.3 *** |
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| α-SMA | 5′-TGACCCAGATTATGTTTGAGACC-3′ | 5′-CCAGAGTCCAGCACAATACCA-3′ |
| COL1-α1 | 5′-GAGCGGAGAGTACTGGATCG-3′ | 5′-GTTCGGGCTGATGTACCAGT-3′ |
| TGF-β1 | 5′-CGCCATCTATGAGAAAACC-3′ | 5′-GTAACGCCAGGAATTGT-3′ |
| TIMP-1 | 5′-GGAAAGCCTCTGTGGATATG-3′ | 5′-AACAGGGAAACACTGTGC-3′ |
| TIMP-2 | 5′-TTCCGGGAATGACATCTATGG-3′ | 5′-GGGCCGTGTAGATAAACTCGAT-3′ |
| MMP-2 | 5′-GCTGATACTGACACTGGTACTG-3′ | 5′-CAATCTTTTCTGGGAGCTC-3′ |
| MMP-9 | 5′-GGAACTCACACGACATCTTCCA-3′ | 5′-GAAACTCACACGCCAGAAGAATTT-3′ |
| Smad-2 | 5′-ATGTCGTCCATCTTGCCATT-3′ | 5′-ATTCTGCTCTCCACCACCTG-3′ |
| Smad-3 | 5′-GTAGAGACGCCAGTTCTACC-3′ | 5′-GGTTTGGAGAACCTGCGTCCAT-3′ |
| Smad-7 | 5′-CAAGAGGCTGTGTTGCTGTG-3′ | 5′-TGGGTATCTGGAGTAAGGAGGA-3′ |
| Cyclophilin | 5′-CCATCG TGTCATCAAGGACTTCAT-3′ | 5′-CTTGCCATCCAGCCAGGAGGTCTT-3′ |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhao, Z.; Yan, Y.; Qiang, X.; Zhou, C.; Li, R.; Chen, H.; Zhang, Y. Demethyleneberberine Protects against Hepatic Fibrosis in Mice by Modulating NF-κB Signaling. Int. J. Mol. Sci. 2016, 17, 1036. https://doi.org/10.3390/ijms17071036
Wang Y, Zhao Z, Yan Y, Qiang X, Zhou C, Li R, Chen H, Zhang Y. Demethyleneberberine Protects against Hepatic Fibrosis in Mice by Modulating NF-κB Signaling. International Journal of Molecular Sciences. 2016; 17(7):1036. https://doi.org/10.3390/ijms17071036
Chicago/Turabian StyleWang, Yongchen, Zheng Zhao, Yan Yan, Xiaoyan Qiang, Cuisong Zhou, Ruiyan Li, Huan Chen, and Yubin Zhang. 2016. "Demethyleneberberine Protects against Hepatic Fibrosis in Mice by Modulating NF-κB Signaling" International Journal of Molecular Sciences 17, no. 7: 1036. https://doi.org/10.3390/ijms17071036
APA StyleWang, Y., Zhao, Z., Yan, Y., Qiang, X., Zhou, C., Li, R., Chen, H., & Zhang, Y. (2016). Demethyleneberberine Protects against Hepatic Fibrosis in Mice by Modulating NF-κB Signaling. International Journal of Molecular Sciences, 17(7), 1036. https://doi.org/10.3390/ijms17071036