
Replacement of Oxygen by Sulfur in Small Organic Molecules. 3. Theoretical Studies on the Tautomeric Equilibria of the 2OH and 4OH-Substituted Oxazole and Thiazole and the 3OH and 4OH-Substituted Isoxazole and Isothiazole in the Isolated State and in Solution

Abstract
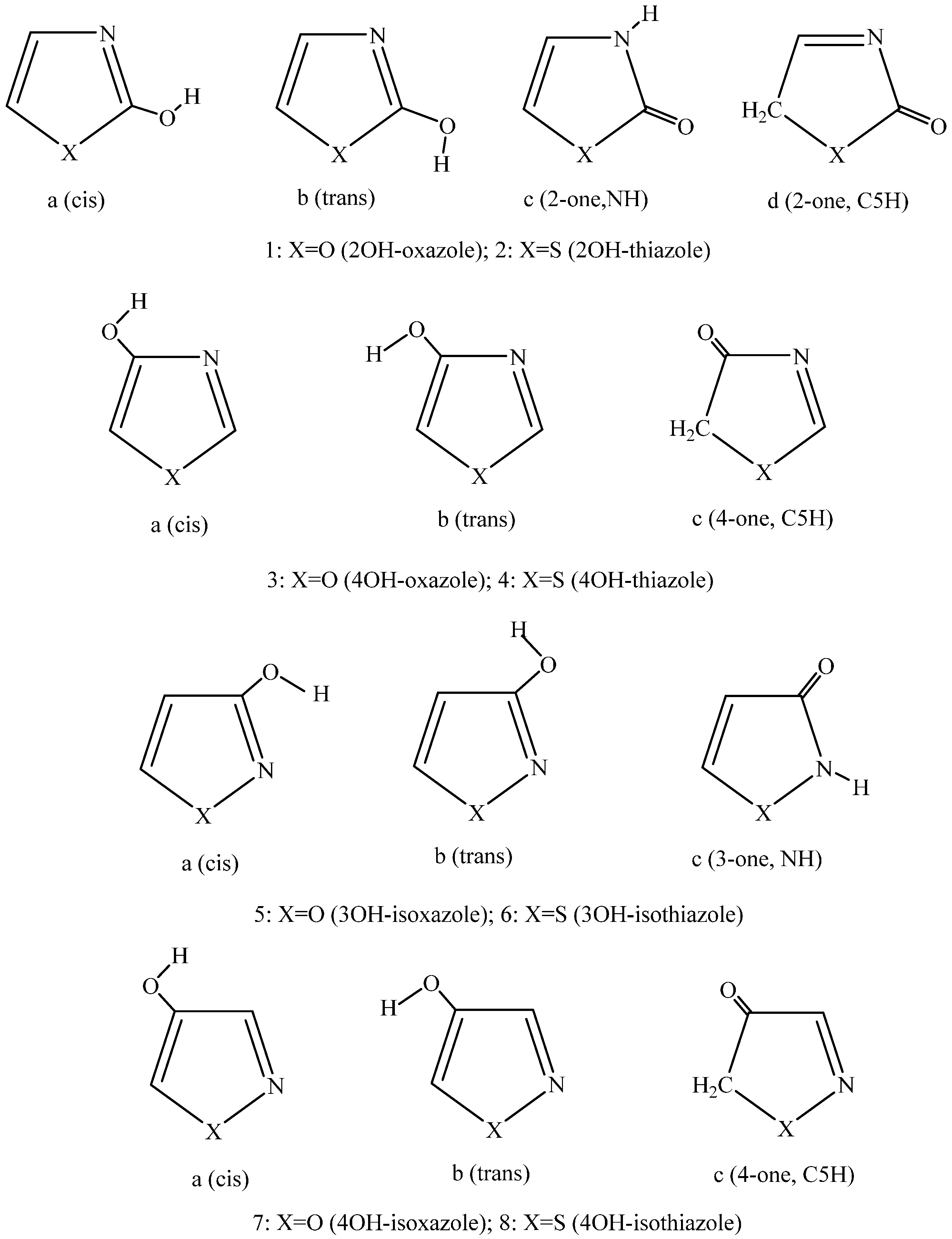
:1. Introduction
2. Results and Discussion
2.1. Methods and Energy Calculations
2.2. Equilibration Mechanism
2.3. Solution Structure Simulations
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Antonov, L. Tautomerism: Methods and Theories; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- Nagy, P.I. Theoretical consideration of in-solution tautomeric equilibria in relation to drug decision. In Tautomerism: Concepts and Applications in Science and Technology; Antonov, L., Ed.; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2016; pp. 113–145. [Google Scholar]
- Martin, Y.C. Let’s not forget tautomers. J. Comput. Aided Mol. Des. 2009, 23, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Hall, C.D.; El-Gendy, B.M.; Draghici, B. Tautomerism in drug discovery. J. Comput. Aided Mol. Des. 2010, 24, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Naray-Szabo, G.; Nagy, P. Electrostatic complementarity in molecular aggregates. Part 9. Protein-ligand complexes. Int. J. Quant. Chem. 1989, 35, 215–221. [Google Scholar] [CrossRef]
- Moriyasu, M.; Kato, A.; Hashimoto, Y. Kinetic studies of fast equilibration by means of high-performance liquid chromatography. Part 11. Keto-enol tautomerism of some β-dicarbonyl compounds. J. Chem. Soc. Perkin Trans. II 1986, 4, 515–520. [Google Scholar] [CrossRef]
- Ishida, T.; Hirata, F.; Kato, S. Thermodynamic analysis of the solvent effect on tautomerization of acetylacetone: An ab initio approach. J. Chem. Phys. 1999, 110, 3938–3945. [Google Scholar] [CrossRef]
- Schlund, S.; Janke, E.M.B.; Weisz, K.; Engels, B. Predicting the tautomeric equilibrium of acetylacetone in solution I. The right answer for the wrong reason? J. Comput. Chem. 2010, 31, 665–670. [Google Scholar] [PubMed]
- Alagona, G.; Ghio, C.; Nagy, P.I. The catalytic effect of water on the keto-enol tautomerism. Pyruvate and acetylacetone: A computational challenge. Phys. Chem. Chem. Phys. 2010, 12, 10173–10188. [Google Scholar] [CrossRef] [PubMed]
- Karelson, M.M.; Katritzky, A.R.; Szafran, M.; Zerner, M.C. A theoretical treatment of solvent effects on the tautomeric equilibria of five-membered rings with two heteroatoms. J. Chem. Soc. Perkin Trans. 1990, 2, 195–201. [Google Scholar] [CrossRef]
- Woodcock, S.; Green, D.V.S.; Vincent, M.A.; Hillier, I.H.; Guest, M.F.; Sherwood, P. Tautomeric equilibria in 3- and 5-hydroxyisoxazole in the gas phase and in aqueous solution: A test of molecular dynamics and continuum models of solvation. J. Chem. Soc. Perkin Trans. 1992, 2, 2151–2154. [Google Scholar] [CrossRef]
- Gould, I.R.; Hillier, I.H. Modelling of tautomeric equilibria of 5-hydroxy-isoxazole in aqueous solution. J. Chem. Soc. Perkin Trans. 1993, 2, 1771–1773. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G. Correlation and solvation effects on heterocyclic equilibria in aqueous solution. J. Am. Chem. Soc. 1993, 115, 8810–8817. [Google Scholar] [CrossRef]
- Parchment, O.G.; Green, D.V.S.; Taylor, P.J.; Hillier, I.H. The prediction of tautomer equilibria in hydrated 3-hydroxypyrazole: a challenge to theory. J. Am. Chem. Soc. 1993, 115, 2352–2356. [Google Scholar] [CrossRef]
- Cao, M.; Teppen, B.J.; Miller, D.M.; Pranata, J.; Schafer, L. Tautomeric equilibria of 3-hydroxypyrazole in the gas phase and in solution: A theoretical study combining ab initio quantum mechanics and Monte Carlo simulation methods. J. Phys. Chem. 1994, 98, 11353–11361. [Google Scholar] [CrossRef]
- Nagy, P.I.; Tejada, F.R.; Messer, W.S., Jr. Theoretical studies of the tautomeric equilibria for five-member N-heterocycles in the gas phase and in solution. J. Phys. Chem. B 2005, 109, 22588–22602. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.I. Theoretical calculations on the conformational/tautomeric equilibria for small molecules in solution. Biochem. Pharmacol. Open Access 2013, S4-001, 1–23. [Google Scholar] [CrossRef]
- Nagy, P.I. In-solution conformational analysis of the XCYCH3 moiety for small esters and ethers with all combinations of X, Y = O, S. Molecules 2013, 18, 8063–8082. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.I. Replacement of oxygen by sulfur in small organic molecules. 2. Theoretical studies on the tautomeric equilibria of the 5OH substituted oxazole, thiazole, isoxazole, and isothiazole in the isolated state and in solution. Curr. Phys. Chem. 2016, in press. [Google Scholar] [CrossRef]
- Barrett, G.C. The chemistry of 1,3-thiazolinone ↔ hydroxy-1,3-thiazole systems. Tetrahedron 1980, 36, 2023–2058. [Google Scholar] [CrossRef]
- Täuscher, E.; Weiß, D.; Beckert, R.; Fabian, J.; Assumpçăo, A.; Görls, H. Classical heterocycles with surprising properties: the 4-hydroxy-1,3-thiazoles. Tetrahedron Lett. 2011, 52, 2292–2294. [Google Scholar]
- Dahlke, E.D.; Cramer, C.J. Prediction of the tautomeric preferences and pKa values for oxyluciferin and its constituent heterocycles. J. Phys. Org. Chem. 2003, 16, 336–347. [Google Scholar] [CrossRef]
- Fast, P.L.; Truchlar, D.G. MC-QCISD: Multi-coefficient correlation method based on quadratic configuration interaction with single and double excitations. J. Phys. Chem. A 2000, 104, 6111–6116. [Google Scholar] [CrossRef]
- Fast, P.L.; Sánchez, M.L.; Truchlar, D.G. Multi-coefficient Gaussian-3 method for calculating potential energy surfaces. Chem. Phys. Lett. 1999, 306, 407–410. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.A. Gaussian basis sets exhibiting systematic convergence to the complete basis set limit. Annu. Rep. Comput. Chem. 2007, 3, 195–206. [Google Scholar]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comp. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. Van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Nagy, P.I.; Alagona, G.; Ghio, C. Theoretical investigation of tautomeric equilibria for isonicotinic acid, 4-pyridone, and acetylacetone in vacuo and in solution. J. Chem. Theory Comput. 2007, 3, 1249–1266. [Google Scholar] [CrossRef] [PubMed]
- McQuarrie, D.A. Statistical Mechanics; University Science Books: Sausalito, CA, USA, 2000. [Google Scholar]
- Jorgensen, W.L. BOSS Version 4.8 Biochemical and Organic Simulation System User’s Manual; Yale University: New Haven, CT, USA, 2007. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Madura, J.D. Quantum and statistical mechanical studies of liquids. 25. Solvation and conformation of methanol in water. J. Am. Chem. Soc. 1983, 105, 1407–1413. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Swenson, C.J. Optimized intermolecular potential functions for amides and peptides. Hydration of amides. J. Am. Chem. Soc. 1985, 107, 1489–1496. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Gao, J. Monte Carlo simulations of the hydration of ammonium and carboxylate ions. J. Phys. Chem. 1986, 90, 2174–2182. [Google Scholar] [CrossRef]
- Lim, D.; Hrovat, D.A.; Borden, W.T.; Jorgensen, W.L. Solvent effects on the ring opening of cyclopropanones to oxyallyls: a combined ab initio and Monte Carlo study. J. Am. Chem. Soc. 1994, 116, 3494–3499. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Rizzo, R.C.; Jorgensen, W.L. OPLS all-atom model for amines: resolution of the amine hydration problem. J. Am. Chem. Soc. 1999, 121, 4827–4836. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Ewald, P.P. Die Berechnung optischer und elektrostatischer Gitterpotentiale. Ann. Phys. 1921, 369, 253–287. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D. Computer Simulations of Liquids; Oxford University Press: Oxford, UK, 1987. [Google Scholar]
- Zwanzig, R.W. High-temperature equation of state by a perturbation method. I. Nonpolar gases. J. Chem. Phys. 1954, 22, 1420–1426. [Google Scholar]
- Jorgensen, W.L.; Ravimohan, C. Monte Carlo simulation of differences in free energies of hydration. J. Chem. Phys. 1985, 83, 3050–3054. [Google Scholar] [CrossRef]
- Nagy, P.I. The syn-anti-equilibrium for the -COOH group reinvestigated. Theoretical conformation analysis for acetic acid in the gas phase and in solution. Comput. Theor. Chem. 2013, 1022, 59–69. [Google Scholar] [CrossRef]
- Nagy, P.I.; Sarver, J.G. Theoretical conformation analysis for chain systems with two conjugated double bonds in the gas phase and in solution. Comput. Theor. Chem. 2014, 1033, 43–51. [Google Scholar] [CrossRef]
- Nagy, P.I. Theoretical in-solution conformational/tautomeric analyses for chain systems with conjugated double bonds involving nitrogen(s). Int. J. Mol. Sci. 2015, 16, 10767–10796. [Google Scholar] [CrossRef] [PubMed]
- Elguero, J.; Marzin, C.; Katritzky, A.R.; Linda, P. Advances in Heterocyclic Chemistry: Tautomerism of Heterocycles, Supplement 1; Academic Press: New York, NY, USA, 1976; pp. 300–303. [Google Scholar]
- Katritzky, A.R. Handbook of Heterocyclic Chemistry; Pergamon Press: Oxford, UK, 1985; pp. 121–123. [Google Scholar]
- De Sarlo, F.; Dini, G.; Lacrimini, P. lsoxazolin 5-one. J. Chem. Soc. C 1971, 86–89. [Google Scholar] [CrossRef]
- Stocks, I.D.H.; Waite, J.A.; Wooldridge, K.R.H. Isothiazoles. Part XIV. 5-alkoxy- and 5-hydroxyisothiazoles. J. Chem. Soc. C 1971, 1314–1317. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Putz, M.V.; Russo, N.; Sicilia, E. On the applicability of the HSAB principle through the use of improved computational schemes for chemical hardness evaluation. J. Comput. Chem. Soc. 2004, 25, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Putz, M.V. Maximum hardness index of quantum acid-base bonding. MATCH Commun. Math. Comput. Chem. 2008, 60, 845–868. [Google Scholar]
- Putz, M.V. Compactness aromaticity of atoms in molecules. Int. J. Mol. Sci. 2010, 11, 1269–1310. [Google Scholar] [CrossRef] [PubMed]
- Cramer, C.C. Essentials of Computational Chemistry: Theories and Models, 2nd ed.; John Wiley & Sons: Chichester, UK, 2004; pp. 85–86. [Google Scholar]
- Nagy, P.I. Are the intramolecular O-H…F and O-H…Cl hydrogen bonds maintained in solution? A theoretical study. J. Phys. Chem. A 2013, 117, 2812–2826. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gas | CH2Cl2 | Water | |||||
|---|---|---|---|---|---|---|---|
| ΔGgint | ΔGsint | ΔGsolv | ΔGstot | ΔGsint | ΔGsolv | ΔGstot | |
| 2OH oxazole | |||||||
| 2OH, trans | 7.8 | 10.0 | −6.6 | 3.4 | 11.2 | −9.1 | 2.1 |
| 2-one, NH | −52.0 | −45.4 | −17.0 | −62.4 | −42.7 | −21.9 | −64.6 |
| 2-one, C5H | −23.6 | −16.8 | −20.6 | −37.4 | −13.4 | −26.8 | −40.2 |
| 2OH thiazole | |||||||
| 2OH, trans | 12.6 | 17.1 | −11.2 | 5.9 | 18.8 | −11.5 | 7.3 |
| 2-one, NH | −45.9 | −40.7 | −14.2 | −54.9 | −38.2 | −14.7 | −52.9 |
| 2-one, C5H | −9.1 | −1.8 | −17.8 | −19.6 | 1.6 | −19.8 | −18.2 |
| 4OH oxazole | |||||||
| 4OH, trans | 11.5 | 15.1 | −10.8 | 4.3 | 16.9 | −14.4 | 2.5 |
| 4-one, C5H | −29.5 | −23.6 | −16.3 | −39.9 | −21.0 | −20.8 | −41.8 |
| 4OH thiazole | |||||||
| 4OH, trans | 11.9 | 16.2 | −11.6 | 4.6 | 18.3 | −15.6 | 2.7 |
| 4-one, C5H | −8.0 | −1.7 | −16.5 | −18.2 | 1.3 | −21.8 | −20.5 |
| Gas | CH2Cl2 | Water | |||||
|---|---|---|---|---|---|---|---|
| ΔGgint | ΔGsint | ΔGsolv | ΔGstot | ΔGsint | ΔGsolv | ΔGstot | |
| 3OH isoxazole | |||||||
| 3OH, trans | 13.5 | 17.9 | −12.7 | 5.2 | 20.1 | −17.2 | 2.9 |
| 3-one, NH | 4.6 b | 9.8 | −11.2 | −3.1 b | 11.3 | −14.3 | −4.7 b |
| 3OH isothiazole | |||||||
| 3OH, trans | 14.9 | 19.3 | −12.9 | 6.4 | 21.6 | −17.5 | 4.1 |
| 3-one, NH | 4.6 | 13.6 | −19.2 | −5.6 | 16.4 | −24.7 | −8.2 |
| 4OH isoxazole | |||||||
| 4OH, trans | 0.0 | 0.1 | −0.2 | −0.1 | 0.1 | −0.2 | −0.1 |
| 4-one, C5H | 1.9 | −0.6 | 6.6 | 6.0 | −1.6 | 9.2 | 7.6 |
| 4OH isothiazole | |||||||
| 4OH, trans | −0.4 | 0.8 | −1.1 | −0.3 | 1.0 | −1.6 | −0.6 |
| 4-one, C5H | 12.9 | 12.7 | 3.3 | 15.9 | 12.3 | 4.8 | 17.1 |
| CH2Cl2 | Water | Exp b,c | |
|---|---|---|---|
| 2OH | |||
| oxazole | 2-one, NH | 2-one, NH | 2-one, NH |
| thiazole | 2-one, NH | 2-one, NH | 2-one, NH |
| 3OH | |||
| isoxazole | 3-one, NH/3OH cis | 3-one, NH | Mainly 3OH in all media |
| isothiazole | 3-one, NH | 3-one, NH | 3OH |
| 4OH | |||
| oxazole | 4-one, C5H | 4-one, C5H | 4-one, C5H |
| thiazole | 4-one, C5H | 4-one, C5H | 4OH and 4-one, C5H no ratio provided |
| isoxazole | 4OH, trans/4OH, cis | 4OH, trans/4OH, cis | 4OH |
| isothiazole | 4OH, trans/4OH, cis | 4OH, trans/4OH, cis | 4OH |
| 5OH d | |||
| oxazole | 5-one, C4H | 5-one, C4H | 5-one, C4H (in crystal) |
| thiazole | 5-one, C4H | 5-one, C4H | |
| isoxazole | 5-one, C4H/5-one, NH | 5-one, C4H/5-one, NH | 5-one, C4H in CH3Cl3, NH/CH 55:45 in water e |
| isothiazole | 5-one, NH | 5-one, NH | 5-one, NH f |
| 3CH3,5OH isoxazole | 5-one, C4H | C4H/NH 70:30 in water | |
| 4CH3,5OH isoxazole | 5-one, NH | 5-one, NH |
| CH2Cl2 | Water | |||||
|---|---|---|---|---|---|---|
| PCM | MC | DM | PCM | MC | DM | |
| Oxazole | ||||||
| 1a → 1c | −17.0 | 0.25, 6.21 | −21.9 | −22.4 | 0.35, 6.52 | |
| 1c → 1d | −3.6 | 0.7 | 6.21, 7.35 | −4.9 | 9.1 | 6.52, 7.71 |
| 3b → 3c | −5.5 | 3.35, 5.15 | −6.4 | −7.6 | 3.54, 5.43 | |
| Thiazole | ||||||
| 2a → 2c | −14.2 | 0.58, 5.59 | −14.7 | −23.7 | 0.62, 5.90 | |
| 2c → 2d | −1.8 | 0.3 | 5.59, 7.08 | −5.1 | 8.1 | 5.90, 7.48 |
| 4b → 4c | −4.9 | 3.51, 5.33 | −6.2 | −5.8 | 3.75, 5.70 | |
| Isoxazole | ||||||
| 5a → 5c | −11.2 | −9.9 | 2.31, 4.88 | −14.3 | −17.7 | 2.43, 5.14 |
| 7b → 7c | 6.8 | 4.46, 0.89 | 9.4 | 8.1 | 4.67, 0.92 | |
| Isothiazole | ||||||
| 6a → 6c | −19.2 | 1.73, 5.34 | −24.7 | −40.3 | 1.84, 5.69 | |
| 8b → 8c | 4.4 | −1.9 | 4.27, 2.05 | 6.4 | 5.8 | 4.51, 2.17 |
| Oring/Hw | Sring/Hw | N/Hw | NH/Ow | −O/Hw | OH/Ow | =O/Hw | nHB(E) | |
|---|---|---|---|---|---|---|---|---|
| Oxazole | ||||||||
| 1a | --- b | 1.4 | 0.6 | 1.0 | 2.1(−14.6) | |||
| 1c | 0.3 c | 1.1 | 2.3 | 2.8(−16.7) | ||||
| 1d | 0.1 c | 0.6 | 1.9 | 2.1(−16.7 d) | ||||
| 3b | 0.4 | 1.3 | 0.8 e | 1.0 | 2.6(−14.6) | |||
| 3c | 0.1 f | 0.9 | 1.9 | 2.4(−16.7) | ||||
| Thiazole | ||||||||
| 2a | --- b | 1.3 g | 0.7 | 1.0 | 1.7(−14.6) | |||
| 2c | --- b | 1.0 | 2.1 | 2.7(−16.7) | ||||
| 2d | --- b | 1.0 | 1.9 | 2.2(−16.7 d) | ||||
| 4b | --- b | 1.6 | 0.9 | 1.0 | 2.7(−14.6) | |||
| 4c | --- b | 1.0 | 2.1 | 2.8(−14.6) | ||||
| Isoxazole | ||||||||
| 5a | 1.9 h | 2.0 i | 0.7 | 1.0 | 2.5(−14.6) | |||
| 5c | --- b | 1.0 | 2.4 | 3.2(−16.7) | ||||
| 7b | 1.1 | 1.7 | 0.8 | 1.0 | 2.2(−16.7) | |||
| 7c | 0.7 j | 1.1 | 1.5 | 2.2(−12.6 d) | ||||
| Isothiazole | ||||||||
| 6a | (0.2) k | 1.5 g | 0.9 | 1.0 | 2.8(−10.5) | |||
| 6c | --- b | 1.0 | 2.6 | 3.5(−16.7) | ||||
| 8b | --- b | 1.5 | 0.9 | 1.0 | 1.9(−16.7 d) | |||
| 8c | --- b | 0.4 l | 1.7 | 1.3(−16.7) |
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy, P.I. Replacement of Oxygen by Sulfur in Small Organic Molecules. 3. Theoretical Studies on the Tautomeric Equilibria of the 2OH and 4OH-Substituted Oxazole and Thiazole and the 3OH and 4OH-Substituted Isoxazole and Isothiazole in the Isolated State and in Solution. Int. J. Mol. Sci. 2016, 17, 1094. https://doi.org/10.3390/ijms17071094
Nagy PI. Replacement of Oxygen by Sulfur in Small Organic Molecules. 3. Theoretical Studies on the Tautomeric Equilibria of the 2OH and 4OH-Substituted Oxazole and Thiazole and the 3OH and 4OH-Substituted Isoxazole and Isothiazole in the Isolated State and in Solution. International Journal of Molecular Sciences. 2016; 17(7):1094. https://doi.org/10.3390/ijms17071094
Chicago/Turabian StyleNagy, Peter I. 2016. "Replacement of Oxygen by Sulfur in Small Organic Molecules. 3. Theoretical Studies on the Tautomeric Equilibria of the 2OH and 4OH-Substituted Oxazole and Thiazole and the 3OH and 4OH-Substituted Isoxazole and Isothiazole in the Isolated State and in Solution" International Journal of Molecular Sciences 17, no. 7: 1094. https://doi.org/10.3390/ijms17071094
APA StyleNagy, P. I. (2016). Replacement of Oxygen by Sulfur in Small Organic Molecules. 3. Theoretical Studies on the Tautomeric Equilibria of the 2OH and 4OH-Substituted Oxazole and Thiazole and the 3OH and 4OH-Substituted Isoxazole and Isothiazole in the Isolated State and in Solution. International Journal of Molecular Sciences, 17(7), 1094. https://doi.org/10.3390/ijms17071094