
The Potential Role of Kallistatin in the Development of Abdominal Aortic Aneurysm



Abstract
:
1. Introduction
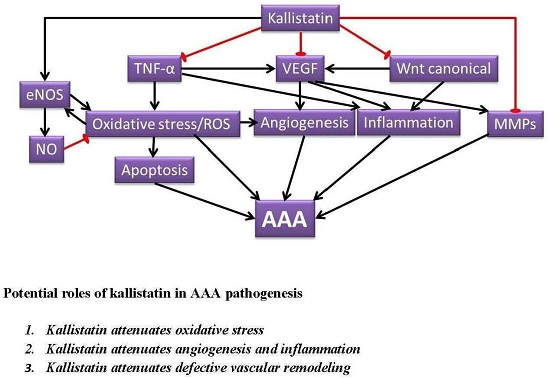
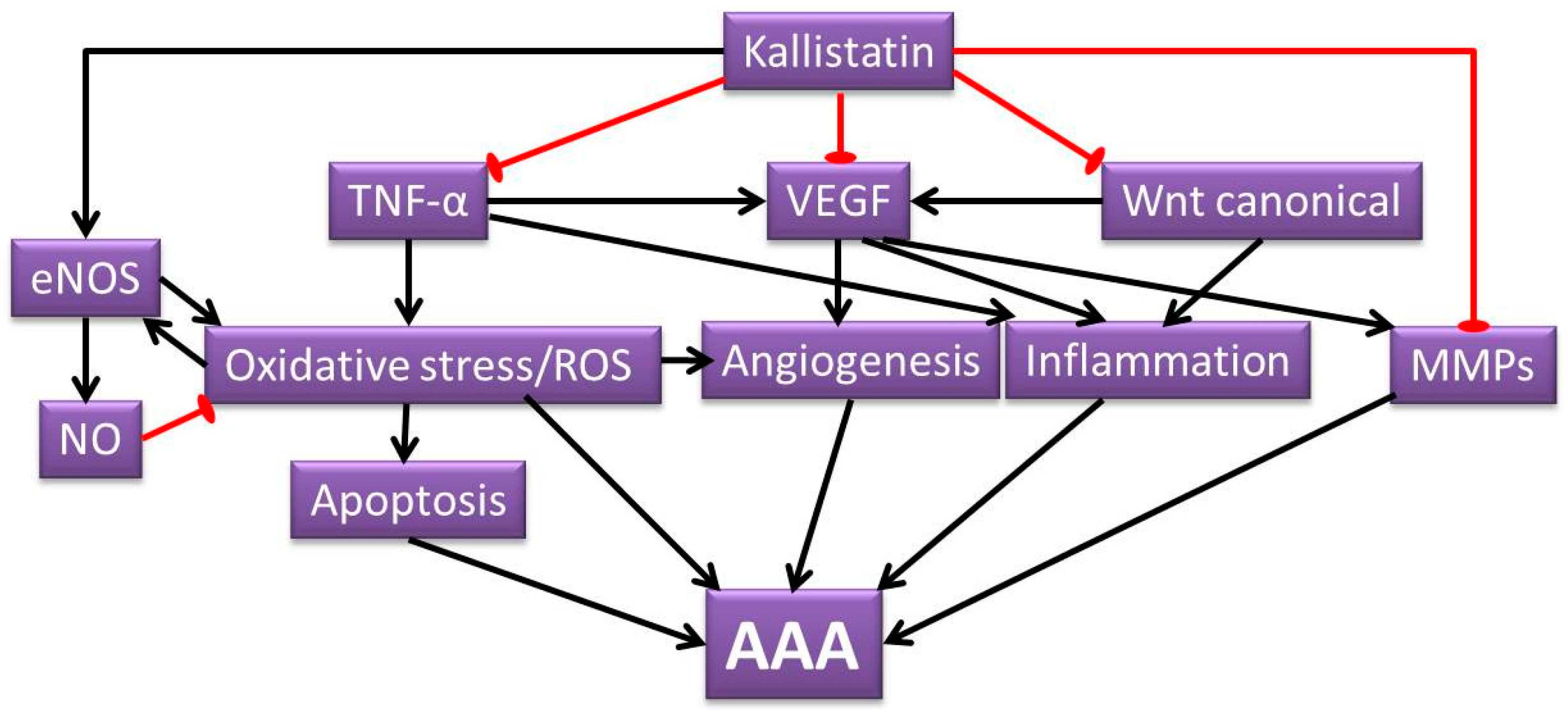
2. Potential Roles of Kallistatin in AAA Pathogenesis
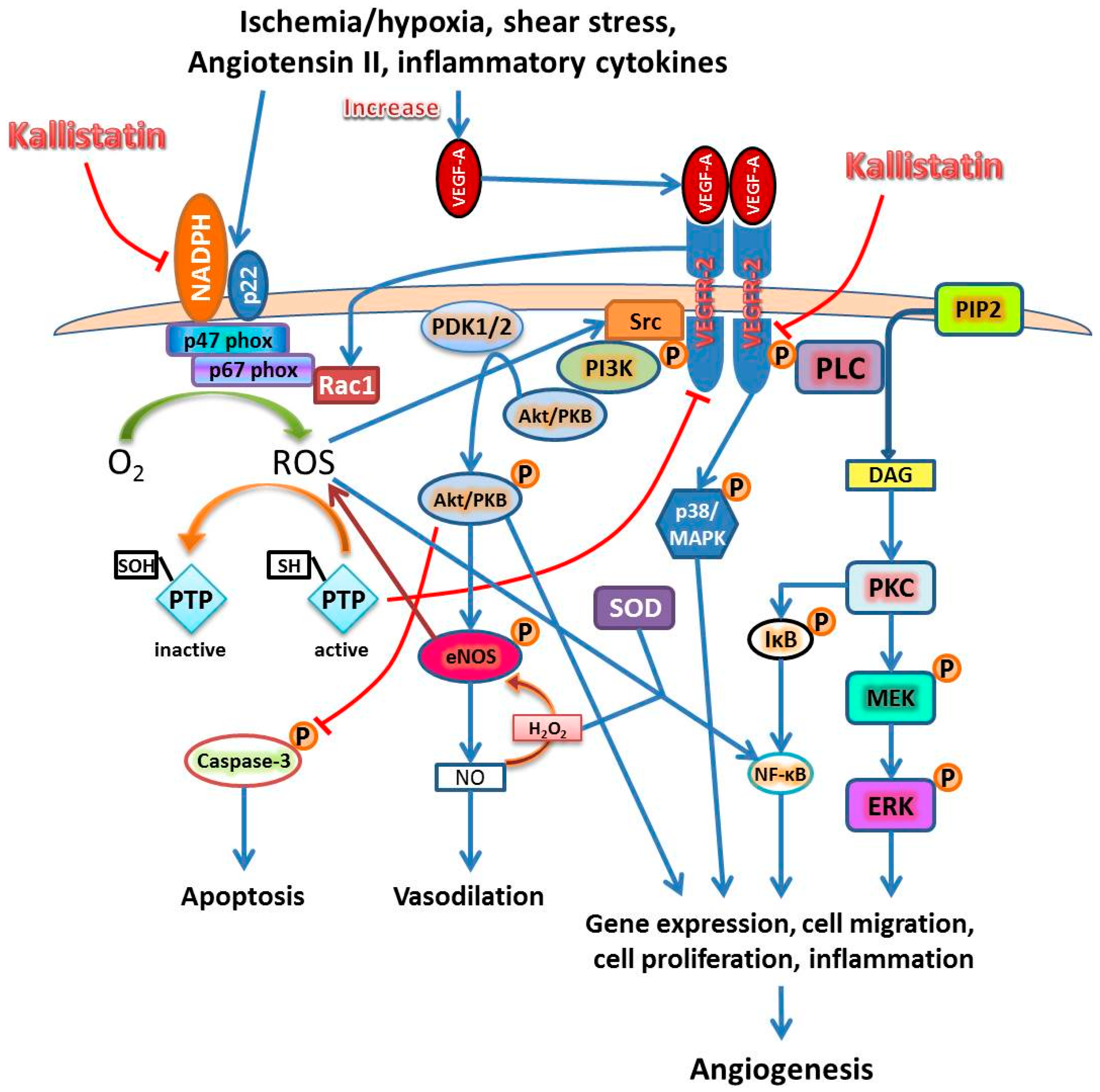
2.1. Kallistatin Attenuates Oxidative Stress
2.2. Kallistatin Attenuates Angiogenesis and Inflammation
2.3. Kallistatin Attenuates Defective Vascular Remodeling
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviation
| AAA | abdominal aortic aneurysm |
| eNOS | endothelial nitric oxide synthase |
| MMPs | matrix metalloproteinases |
| NO | nitric oxide |
| ROS | reactive oxygen species |
| TNF-α | tumor necrosis factor alpha |
| VEGF | vascular endothelial growth factor |
References
- Sakalihasan, N.; Limet, R.; Defawe, O.D. Abdominal aortic aneurysm. Lancet 2005, 365, 1577–1589. [Google Scholar] [CrossRef]
- Moxon, J.V.; Parr, A.; Emeto, T.I.; Walker, P.; Norman, P.E.; Golledge, J. Diagnosis and monitoring of abdominal aortic aneurysm: Current status and future prospects. Curr. Probl. Cardiol. 2010, 35, 512–548. [Google Scholar] [CrossRef] [PubMed]
- Gillum, R.F. Epidemiology of aortic aneurysm in the united states. J. Clin. Epidemiol. 1995, 48, 1289–1298. [Google Scholar] [CrossRef]
- Kniemeyer, H.W.; Kessler, T.; Reber, P.U.; Ris, H.B.; Hakki, H.; Widmer, M.K. Treatment of ruptured abdominal aortic aneurysm, a permanent challenge or a waste of resources? Prediction of outcome using a multi-organ-dysfunction score. Eur. J. Vasc. Endovasc. Surg. 2000, 19, 190–196. [Google Scholar] [CrossRef] [PubMed]
- The UK small aneurysm trial participants. Mortality results for randomised controlled trial of early elective surgery or ultrasonographic surveillance for small abdominal aortic aneurysms. Lancet 1998, 352, 1649–1655. [Google Scholar]
- Melrose, J.; Whitelock, J.; Xu, Q.; Ghosh, P. Pathogenesis of abdominal aortic aneurysms: Possible role of differential production of proteoglycans by smooth muscle cells. J. Vasc. Surg. 1998, 28, 676–686. [Google Scholar] [CrossRef]
- Baxter, B.T.; McGee, G.S.; Shively, V.P.; Drummond, I.A.; Dixit, S.N.; Yamauchi, M.; Pearce, W.H. Elastin content, cross-links, and mrna in normal and aneurysmal human aorta. J. Vasc. Surg. 1992, 16, 192–200. [Google Scholar] [CrossRef]
- Sakalihasan, N.; Heyeres, A.; Nusgens, B.V.; Limet, R.; Lapiere, C.M. Modifications of the extracellular matrix of aneurysmal abdominal aortas as a function of their size. Eur. J. Vasc. Surg. 1993, 7, 633–637. [Google Scholar] [CrossRef]
- Hellenthal, F.A.; Buurman, W.A.; Wodzig, W.K.; Schurink, G.W. Biomarkers of AAA progression. Part 1: Extracellular matrix degeneration. Nat. Rev. Cardiol. 2009, 6, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.E.; Kunkel, S.L.; Pearce, W.H.; Shah, M.R.; Parikh, D.; Evanoff, H.L.; Haines, G.K.; Burdick, M.D.; Strieter, R.M. Enhanced production of the chemotactic cytokines interleukin-8 and monocyte chemoattractant protein-1 in human abdominal aortic aneurysms. Am. J. Pathol. 1993, 142, 1423–1431. [Google Scholar] [PubMed]
- Newman, K.M.; Jean-Claude, J.; Li, H.; Ramey, W.G.; Tilson, M.D. Cytokines that activate proteolysis are increased in abdominal aortic aneurysms. Circulation 1994, 90, II224–II227. [Google Scholar] [PubMed]
- Shah, P.K. Inflammation, metalloproteinases, and increased proteolysis: An emerging pathophysiological paradigm in aortic aneurysm. Circulation 1997, 96, 2115–2117. [Google Scholar] [CrossRef] [PubMed]
- Walton, L.J.; Franklin, I.J.; Bayston, T.; Brown, L.C.; Greenhalgh, R.M.; Taylor, G.W.; Powell, J.T. Inhibition of prostaglandin e2 synthesis in abdominal aortic aneurysms: Implications for smooth muscle cell viability, inflammatory processes, and the expansion of abdominal aortic aneurysms. Circulation 1999, 100, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Jagadesham, V.P.; Scott, D.J.; Carding, S.R. Abdominal aortic aneurysms: An autoimmune disease? Trends Mol. Med. 2008, 14, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Choke, E.; Thompson, M.M.; Dawson, J.; Wilson, W.R.; Sayed, S.; Loftus, I.M.; Cockerill, G.W. Abdominal aortic aneurysm rupture is associated with increased medial neovascularization and overexpression of proangiogenic cytokines. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2077–2082. [Google Scholar] [CrossRef] [PubMed]
- Guzik, B.; Sagan, A.; Ludew, D.; Mrowiecki, W.; Chwala, M.; Bujak-Gizycka, B.; Filip, G.; Grudzien, G.; Kapelak, B.; Zmudka, K.; et al. Mechanisms of oxidative stress in human aortic aneurysms—Association with clinical risk factors for atherosclerosis and disease severity. Int. J. Cardiol. 2013, 168, 2389–2396. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Schmaier, A.; Chen, L.M.; Yang, Z.; Chao, L. Kallistatin, a novel human tissue kallikrein inhibitor: Levels in body fluids, blood cells, and tissues in health and disease. J. Lab. Clin. Med. 1996, 127, 612–620. [Google Scholar] [CrossRef]
- Chao, J.; Tillman, D.M.; Wang, M.Y.; Margolius, H.S.; Chao, L. Identification of a new tissue-kallikrein-binding protein. Biochem. J. 1986, 239, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Stallone, J.N.; Liang, Y.M.; Chen, L.M.; Wang, D.Z.; Chao, L. Kallistatin is a potent new vasodilator. J. Clin. Investig. 1997, 100, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Wolf, W.C.; Harley, R.A.; Sluce, D.; Chao, L.; Chao, J. Localization and expression of tissue kallikrein and kallistatin in human blood vessels. J. Histochem. Cytochem. 1999, 47, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.X.; King, L.P.; Yang, Z.; Crouch, R.K.; Chao, L.; Chao, J. Kallistatin in human ocular tissues: Reduced levels in vitreous fluids from patients with diabetic retinopathy. Curr. Eye Res. 1996, 15, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Chao, J.; Kotak, I.; Guo, D.; Parikh, S.J.; Bhagatwala, J.; Dong, Y.; Patel, S.Y.; Houk, C.; Chao, L.; et al. Plasma kallistatin is associated with adiposity and cardiometabolic risk in apparently healthy african american adolescents. Metabolism 2013, 62, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.L.; Tsai, C.Y.; Luo, Y.H.; Kuo, C.F.; Lin, W.C.; Chang, Y.T.; Wu, J.J.; Chuang, W.J.; Liu, C.C.; Chao, L.; et al. Kallistatin modulates immune cells and confers anti-inflammatory response to protect mice from group a streptococcal infection. Antimicrob. Agents Chemother. 2013, 57, 5366–5372. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Gao, L.; Shen, B.; Chao, L.; Chao, J. Kallistatin inhibits vascular inflammation by antagonizing tumor necrosis factor-α-induced nuclear factor κB activation. Hypertension 2010, 56, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bledsoe, G.; Hagiwara, M.; Shen, B.; Chao, L.; Chao, J. Depletion of endogenous kallistatin exacerbates renal and cardiovascular oxidative stress, inflammation, and organ remodeling. Am. J. Physiol. Ren. Physiol. 2012, 303, F1230–F1238. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.R.; Chen, S.Y.; Wu, C.L.; Liu, M.F.; Jin, Y.T.; Chao, L.; Chao, J. Prophylactic adenovirus-mediated human kallistatin gene therapy suppresses rat arthritis by inhibiting angiogenesis and inflammation. Arthritis Rheumatol. 2005, 52, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Gao, L.; Hsu, Y.T.; Bledsoe, G.; Hagiwara, M.; Chao, L.; Chao, J. Kallistatin attenuates endothelial apoptosis through inhibition of oxidative stress and activation of Akt-eNOS signaling. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1419–H1427. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.F.; Huang, X.P.; Xiao, G.Q.; Yang, H.Y.; Lin, J.S.; Diao, Y. Kallistatin, a novel anti-angiogenesis agent, inhibits angiogenesis via inhibition of the NF-κB signaling pathway. Biomed. Pharmacother. 2014, 68, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.F.; Yang, H.Y.; Xing, Y.M.; Lin, J.S.; Diao, Y. Recombinant human kallistatin inhibits angiogenesis by blocking VEGF signaling pathway. J. Cell. Biochem. 2014, 115, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.M.; Chao, L.; Chao, J. Adenovirus-mediated delivery of human kallistatin gene reduces blood pressure of spontaneously hypertensive rats. Hum. Gene Ther. 1997, 8, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.M.; Ma, J.; Liang, Y.M.; Chao, L.; Chao, J. Tissue kallikrein-binding protein reduces blood pressure in transgenic mice. J. Biol. Chem. 1996, 271, 27590–27594. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.C.; Chao, L.; Pimenta, D.C.; Bledsoe, G.; Juliano, L.; Chao, J. Identification of a major heparin-binding site in kallistatin. J. Biol. Chem. 2001, 276, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Miao, R.Q.; Agata, J.; Chao, L.; Chao, J. Kallistatin is a new inhibitor of angiogenesis and tumor growth. Blood 2002, 100, 3245–3252. [Google Scholar] [CrossRef] [PubMed]
- Miao, R.Q.; Chen, V.; Chao, L.; Chao, J. Structural elements of kallistatin required for inhibition of angiogenesis. Am. J. Physiol. Cell Physiol. 2003, 284, C1604–C1613. [Google Scholar] [PubMed]
- Watanabe, A.; Ichiki, T.; Sankoda, C.; Takahara, Y.; Ikeda, J.; Inoue, E.; Tokunou, T.; Kitamoto, S.; Sunagawa, K. Suppression of abdominal aortic aneurysm formation by inhibition of prolyl hydroxylase domain protein through attenuation of inflammation and extracellular matrix disruption. Clin. Sci. 2014, 126, 671–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Cheng, X.; Li, H.; Qiu, F.; Yang, N.; Wang, B.; Lu, H.; Wu, H.; Shen, Y.; Wang, Y.; et al. Quercetin reduces oxidative stress and inhibits activation of c-Jun N-terminal kinase/activator protein1 signaling in an experimental mouse model of abdominal aortic aneurysm. Mol. Med. Rep. 2014, 9, 435–442. [Google Scholar] [PubMed]
- Emeto, T.I.; Moxon, J.V.; Au, M.; Golledge, J. Oxidative stress and abdominal aortic aneurysm: Potential treatment targets. Clin. Sci. 2016, 130, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Miwa, K.; Nakashima, H.; Aoki, M.; Miyake, T.; Kawasaki, T.; Iwai, M.; Oishi, M.; Kataoka, K.; Ohgi, S.; Ogihara, T.; et al. Inhibition of ets, an essential transcription factor for angiogenesis, to prevent the development of abdominal aortic aneurysm in a rat model. Gene Ther. 2005, 12, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J.; Norman, P.E. Current status of medical management for abdominal aortic aneurysm. Atherosclerosis 2011, 217, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Golledge, A.L.; Walker, P.; Norman, P.E.; Golledge, J. A systematic review of studies examining inflammation associated cytokines in human abdominal aortic aneurysm samples. Dis. Mark. 2009, 26, 181–188. [Google Scholar] [CrossRef]
- Grell, M.; Douni, E.; Wajant, H.; Lohden, M.; Clauss, M.; Maxeiner, B.; Georgopoulos, S.; Lesslauer, W.; Kollias, G.; Pfizenmaier, K.; et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 1995, 83, 793–802. [Google Scholar] [CrossRef]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Smith, R.S., Jr.; Hsu, Y.T.; Chao, L.; Chao, J. Kruppel-like factor 4 is a novel mediator of kallistatin in inhibiting endothelial inflammation via increased endothelial nitric-oxide synthase expression. J. Biol. Chem. 2009, 284, 35471–35478. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Li, P.; Zhang, J.; Hagiwara, M.; Shen, B.; Bledsoe, G.; Chang, E.; Chao, L.; Chao, J. Novel role of kallistatin in vascular repair by promoting mobility, viability, and function of endothelial progenitor cells. J. Am. Heart Assoc. 2014, 3, e001194. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Hagiwara, M.; Yao, Y.Y.; Chao, L.; Chao, J. Salutary effect of kallistatin in salt-induced renal injury, inflammation, and fibrosis via antioxidative stress. Hypertension 2008, 51, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M. Redox signaling in angiogenesis: Role of nadph oxidase. Cardiovasc. Res. 2006, 71, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Tang, Y.; Fukai, T.; Dikalov, S.I.; Ma, Y.; Fujimoto, M.; Quinn, M.T.; Pagano, P.J.; Johnson, C.; Alexander, R.W. Novel role of gp91phox-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ. Res. 2002, 91, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Abid, M.R.; Spokes, K.C.; Shih, S.C.; Aird, W.C. Nadph oxidase activity selectively modulates vascular endothelial growth factor signaling pathways. J. Biol. Chem. 2007, 282, 35373–35385. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Alexander, R.W. Reactive oxygen species as mediators of angiogenesis signaling: Role of NAD(P)H oxidase. Mol. Cell. Biochem. 2004, 264, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Harfouche, R.; Malak, N.A.; Brandes, R.P.; Karsan, A.; Irani, K.; Hussain, S.N. Roles of reactive oxygen species in angiopoietin-1/tie-2 receptor signaling. FASEB J. 2005, 19, 1728–1730. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.; Madeddu, P.; Wang, C.; Liang, Y.; Chao, L.; Chao, J. Differential regulation of kallikrein, kininogen, and kallikrein-binding protein in arterial hypertensive rats. Am. J. Physiol. 1996, 271, F78–F86. [Google Scholar] [PubMed]
- Shen, B.; Chao, L.; Chao, J. Pivotal role of JNK-dependent FOXO1 activation in downregulation of kallistatin expression by oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1048–H1054. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Yin, H.; Smith, R.S.; Chao, L.; Chao, J. Role of kallistatin in prevention of cardiac remodeling after chronic myocardial infarction. Lab. Investig. 2008, 88, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Yiu, W.H.; Wong, D.W.; Wu, H.J.; Li, R.X.; Yam, I.; Chan, L.Y.; Leung, J.C.; Lan, H.Y.; Lai, K.N.; Tang, S.C. Kallistatin protects against diabetic nephropathy in db/db mice by suppressing AGE-RAGE-induced oxidative stress. Kidney Int. 2015. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Pan, F.; Ge, L.; Zhou, J.; Chen, L.; Zhou, T.; Zong, R.; Xiao, X.; Dong, N.; Yang, M.; et al. SERPINA3K plays antioxidant roles in cultured pterygial epithelial cells through regulating ROS system. PLoS ONE 2014, 9, e108859. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, S.; Yan, L.; Madara, P.; Del, P.C.A.; Wesley, R.A.; Danner, R.L. Superoxide production and reactive oxygen species signaling by endothelial nitric-oxide synthase. J. Biol. Chem. 2000, 275, 16899–16903. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Cao, P.; Goeddel, D.V. TNF-induced recruitment and activation of the IKK complex require Cdc37 and Hsp90. Mol. Cell 2002, 9, 401–410. [Google Scholar] [CrossRef]
- Giraudo, E.; Primo, L.; Audero, E.; Gerber, H.P.; Koolwijk, P.; Soker, S.; Klagsbrun, M.; Ferrara, N.; Bussolino, F. Tumor necrosis factor-α regulates expression of vascular endothelial growth factor receptor-2 and of its co-receptor neuropilin-1 in human vascular endothelial cells. J. Biol. Chem. 1998, 273, 22128–22135. [Google Scholar] [CrossRef] [PubMed]
- Guadagni, F.; Ferroni, P.; Palmirotta, R.; Portarena, I.; Formica, V.; Roselli, M. Review. TNF/VEGF cross-talk in chronic inflammation-related cancer initiation and progression: An early target in anticancer therapeutic strategy. In Vivo 2007, 21, 147–161. [Google Scholar] [PubMed]
- Ristimaki, A.; Narko, K.; Enholm, B.; Joukov, V.; Alitalo, K. Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J. Biol. Chem. 1998, 273, 8413–8418. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Suzuki, S.; Kawasaki, N.; Nakano, H.; Okazaki, T.; Chino, A.; Doi, T.; Saiki, I. Tumor necrosis factor-α-induced IKK phosphorylation of NF-κB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J. Biol. Chem. 2003, 278, 36916–36923. [Google Scholar] [CrossRef] [PubMed]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.M.; Jones, L.; Nasim, A.; Sayers, R.D.; Bell, P.R. Angiogenesis in abdominal aortic aneurysms. Eur. J. Vasc. Endovasc. Surg. 1996, 11, 464–469. [Google Scholar] [CrossRef]
- Majno, G. Chronic inflammation: Links with angiogenesis and wound healing. Am. J. Pathol. 1998, 153, 1035–1039. [Google Scholar] [CrossRef]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Bagli, E.; Xagorari, A.; Papetropoulos, A.; Murphy, C.; Fotsis, T. Angiogenesis in inflammation. Autoimmun. Rev. 2004, 3, S26. [Google Scholar] [PubMed]
- Firestein, G.S. Starving the synovium: Angiogenesis and inflammation in rheumatoid arthritis. J. Clin. Investig. 1999, 103, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Szekanecz, Z.; Koch, A.E. Vascular endothelium and immune responses: Implications for inflammation and angiogenesis. Rheum. Dis. Clin. N. Am. 2004, 30, 97–114. [Google Scholar] [CrossRef]
- Jackson, J.R.; Seed, M.P.; Kircher, C.H.; Willoughby, D.A.; Winkler, J.D. The codependence of angiogenesis and chronic inflammation. FASEB J. 1997, 11, 457–465. [Google Scholar] [PubMed]
- Choke, E.; Cockerill, G.W.; Dawson, J.; Wilson, R.W.; Jones, A.; Loftus, I.M.; Thompson, M.M. Increased angiogenesis at the site of abdominal aortic aneurysm rupture. Ann. N. Y. Acad. Sci. 2006, 1085, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of vegf and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F.; Detmar, M.; Claffey, K.P.; Nagy, J.A.; van de Water, L.; Senger, D.R. Vascular permeability factor/vascular endothelial growth factor: An important mediator of angiogenesis in malignancy and inflammation. Int. Arch. Allergy Immunol. 1995, 107, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C. The involvement of VEGF in endothelial permeability: A target for anti-inflammatory therapy. Curr. Opin. Investig. Drugs 2005, 6, 1124–1130. [Google Scholar] [PubMed]
- Taimeh, Z.; Loughran, J.; Birks, E.J.; Bolli, R. Vascular endothelial growth factor in heart failure. Nat. Rev. Cardiol. 2013, 10, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Abid, M.R.; Tsai, J.C.; Spokes, K.C.; Deshpande, S.S.; Irani, K.; Aird, W.C. Vascular endothelial growth factor induces manganese-superoxide dismutase expression in endothelial cells by a Rac1-regulated NADPH oxidase-dependent mechanism. FASEB J. 2001, 15, 2548–2550. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P.; Cirri, P. Redox regulation of protein tyrosine phosphatases during receptor tyrosine kinase signal transduction. Trends Biochem. Sci. 2003, 28, 509–514. [Google Scholar] [CrossRef]
- Colavitti, R.; Pani, G.; Bedogni, B.; Anzevino, R.; Borrello, S.; Waltenberger, J.; Galeotti, T. Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. J. Biol. Chem. 2002, 277, 3101–3108. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [PubMed]
- Downward, J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr. Opin. Cell Biol. 1998, 10, 262–267. [Google Scholar] [CrossRef]
- Fulton, D.; Gratton, J.P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [PubMed]
- Gee, E.; Milkiewicz, M.; Haas, T.L. P38 MAPK activity is stimulated by vascular endothelial growth factor receptor 2 activation and is essential for shear stress-induced angiogenesis. J. Cell. Physiol. 2010, 222, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.P.; McMurtrey, A.; Kowalski, J.; Yan, M.; Keyt, B.A.; Dixit, V.; Ferrara, N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. J. Biol. Chem. 1998, 273, 30336–30343. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A. Akt signaling: Linking membrane events to life and death decisions. Science 1997, 275, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Sankar, S.; Lin, C.; Kontos, C.D.; Schroff, A.D.; Cha, E.H.; Feng, S.M.; Li, S.F.; Yu, Z.; van Etten, R.L.; et al. HCPTPA, a protein tyrosine phosphatase that regulates vascular endothelial growth factor receptor-mediated signal transduction and biological activity. J. Biol. Chem. 1999, 274, 38183–38188. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Moon, S.O.; Kim, S.H.; Kim, H.J.; Koh, Y.S.; Koh, G.Y. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa b activation in endothelial cells. J. Biol. Chem. 2001, 276, 7614–7620. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Cheng, G.; Arnold, R.S.; Edens, W.A. Novel homologs of gp91phox. Trends Biochem. Sci. 2000, 25, 459–461. [Google Scholar] [CrossRef]
- Lamers, J.M.; de Jonge, H.W.; Panagia, V.; van Heugten, H.A. Receptor-mediated signalling pathways acting through hydrolysis of membrane phospholipids in cardiomyocytes. Cardioscience 1993, 4, 121–131. [Google Scholar] [PubMed]
- Lassegue, B.; Clempus, R.E. Vascular NAD(P)H oxidases: Specific features, expression, and regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R277–R297. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Yen, M.L.; Lin, C.Y.; Kuo, M.L. Inhibition of vascular endothelial growth factor-induced angiogenesis by resveratrol through interruption of Src-dependent vascular endothelial cadherin tyrosine phosphorylation. Mol. Pharmacol. 2003, 64, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Marumo, T.; Schini-Kerth, V.B.; Busse, R. Vascular endothelial growth factor activates nuclear factor-κB and induces monocyte chemoattractant protein-1 in bovine retinal endothelial cells. Diabetes 1999, 48, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Mei, A.; Liu, J.; Kang, X.; Shi, X.; Qian, R.; Chen, S. Nadph oxidase 4 mediates insulin-stimulated HIF-1α and VEGF expression, and angiogenesis in vitro. PLoS ONE 2012, 7, e48393. [Google Scholar] [CrossRef] [PubMed]
- Rottbauer, W.; Just, S.; Wessels, G.; Trano, N.; Most, P.; Katus, H.A.; Fishman, M.C. VEGF-PLCγ1 pathway controls cardiac contractility in the embryonic heart. Genes Dev. 2005, 19, 1624–1634. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, S.; Houle, F.; Landry, J.; Huot, J. P38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene 1997, 15, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Shiojima, I.; Walsh, K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ. Res. 2002, 90, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka-Tojo, M.; Ushio-Fukai, M.; Hilenski, L.; Dikalov, S.I.; Chen, Y.E.; Tojo, T.; Fukai, T.; Fujimoto, M.; Patrushev, N.A.; Wang, N.; et al. IQGAP1, a novel vascular endothelial growth factor receptor binding protein, is involved in reactive oxygen species—Dependent endothelial migration and proliferation. Circ. Res. 2004, 95, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, C.; Peng, S.; Xu, H.; Wright, E.; Zhang, X.; Huo, X.; Cheng, E.; Pham, T.H.; Asanuma, K.; et al. Autocrine VEGF signaling promotes proliferation of neoplastic barrett’s epithelial cells through a PLC-dependent pathway. Gastroenterology 2014, 146, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Reis, M.; Liebner, S. Wnt signaling in the vasculature. Exp. Cell Res. 2013, 319, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Ju, R.; Cirone, P.; Lin, S.; Griesbach, H.; Slusarski, D.C.; Crews, C.M. Activation of the planar cell polarity formin daam1 leads to inhibition of endothelial cell proliferation, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 6906–6911. [Google Scholar] [CrossRef] [PubMed]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y. Roles of Wnt signaling in bone metabolism. Clin. Calcium 2012, 22, 1701–1706. [Google Scholar] [PubMed]
- Liebner, S.; Plate, K.H. Differentiation of the brain vasculature: The answer came blowing by the Wnt. J. Angiogenes Res. 2010, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, B.; McBride, J.D.; Zhou, K.; Lee, K.; Zhou, Y.; Liu, Z.; Ma, J.X. Antiangiogenic and antineuroinflammatory effects of kallistatin through interactions with the canonical Wnt pathway. Diabetes 2013, 62, 4228–4238. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Takahashi, N.; Kobayashi, Y. Roles of Wnt signals in bone resorption during physiological and pathological states. J. Mol. Med. (Berl.) 2013, 91, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, L.M.; Nilsson-Ohman, J.; Zetterqvist, A.V.; Gomez, M.F. Nuclear factor of activated T-cells transcription factors in the vasculature: The good guys or the bad guys? Curr. Opin. Lipidol. 2008, 19, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Ravindranath, A.; Yuen, H.F.; Chan, K.K.; Grills, C.; Fennell, D.A.; Lappin, T.R.; El-Tanani, M. Wnt-β-catenin-Tcf-4 signalling-modulated invasiveness is dependent on osteopontin expression in breast cancer. Br. J. Cancer 2011, 105, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene 2006, 25, 7469–7481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, Z.; Li, P.; Bledsoe, G.; Chao, L.; Chao, J. Kallistatin antagonizes Wnt/β-catenin signaling and cancer cell motility via binding to low-density lipoprotein receptor-related protein 6. Mol. Cell. Biochem. 2013, 379, 295–301. [Google Scholar] [CrossRef] [PubMed]
- McBride, J.D.; Jenkins, A.J.; Liu, X.; Zhang, B.; Lee, K.; Berry, W.L.; Janknecht, R.; Griffin, C.T.; Aston, C.E.; Lyons, T.J.; et al. Elevated circulation levels of an antiangiogenic serpin in patients with diabetic microvascular complications impair wound healing through suppression of Wnt signaling. J. Investig. Dermatol. 2014, 134, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Shao, C.; Zhang, S.X.; Dudley, A.; Fant, J.; Ma, J.X. Kallikrein-binding protein inhibits retinal neovascularization and decreases vascular leakage. Diabetologia 2003, 46, 689–698. [Google Scholar] [PubMed]
- Liu, X.; McBride, J.; Zhou, Y.; Liu, Z.; Ma, J.X. Regulation of endothelial progenitor cell release by Wnt signaling in bone marrow. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7386–7394. [Google Scholar] [CrossRef] [PubMed]
- Phng, L.K.; Gerhardt, H. Angiogenesis: A team effort coordinated by notch. Dev. Cell 2009, 16, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Renna, N.F.; de Las Heras, N.; Miatello, R.M. Pathophysiology of vascular remodeling in hypertension. Int. J. Hypertens. 2013, 2013, 808353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, C.S.; Rush, C.M.; Dougan, T.; Jose, R.J.; Biros, E.; Norman, P.E.; Gera, L.; Golledge, J. Modulation of kinin B2 receptor signaling controls aortic dilatation and rupture in the angiotensin II-infused apolipoprotein E-deficient mouse. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Chao, L. Kallistatin in blood pressure regulation transgenic and somatic gene delivery studies. Trends Cardiovasc. Med. 1997, 7, 307–311. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibited Pathology | In Vitro Model | In Vivo Model | References |
|---|---|---|---|
| Oxidative stress/inflammation | Proximal tubular cells, mesangial cells | Dahl-salt sensitive rats | [45] |
| HUVEC | – | [43] | |
| – | Hypertensive rats | [25] | |
| Oxidative stress/apoptosis | Rat and human endo-PC | Deoxycorticosterone acetate salt-hypertensive rats | [44] |
| HUVEC | Rats | [27] |
| Inhibited Pathology | Pathways | In Vitro Model | In Vivo Model | References |
|---|---|---|---|---|
| Retinal neovascularisation/angiogenesis | VEGF | Retinal capillary ECs | Brown Norway rats | [112] |
| Angiogenesis in cancer | TNF-α/VEGF | MCF-7 cells, HUVEC | Fertilized chicken egg | [28] |
| Angiogenesis/inflammation arthritis | TNF-α | HDMEC | Rats | [26] |
| Angiogenesis/Inflammation Diabetic or OIR | Wnt canonical pathway | Retinal cells | Kallistatin transgenic mice with OIR or type I diabetes | [105] |
| Oxygen induced retinopathy/angiogenesis | Wnt canonical pathway | – | Kallistatin transgenic mice, bet-gal mice | [113] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Krishna, S.M.; Golledge, J. The Potential Role of Kallistatin in the Development of Abdominal Aortic Aneurysm. Int. J. Mol. Sci. 2016, 17, 1312. https://doi.org/10.3390/ijms17081312
Li J, Krishna SM, Golledge J. The Potential Role of Kallistatin in the Development of Abdominal Aortic Aneurysm. International Journal of Molecular Sciences. 2016; 17(8):1312. https://doi.org/10.3390/ijms17081312
Chicago/Turabian StyleLi, Jiaze, Smriti Murali Krishna, and Jonathan Golledge. 2016. "The Potential Role of Kallistatin in the Development of Abdominal Aortic Aneurysm" International Journal of Molecular Sciences 17, no. 8: 1312. https://doi.org/10.3390/ijms17081312
APA StyleLi, J., Krishna, S. M., & Golledge, J. (2016). The Potential Role of Kallistatin in the Development of Abdominal Aortic Aneurysm. International Journal of Molecular Sciences, 17(8), 1312. https://doi.org/10.3390/ijms17081312