
Epigenetic Mechanisms in Bone Biology and Osteoporosis: Can They Drive Therapeutic Choices?



Abstract
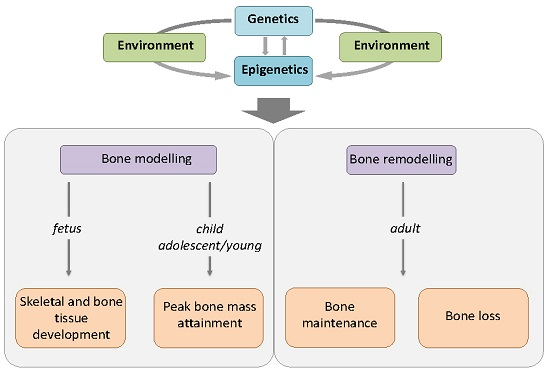
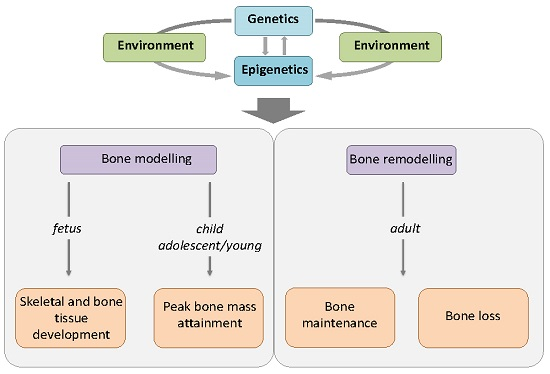
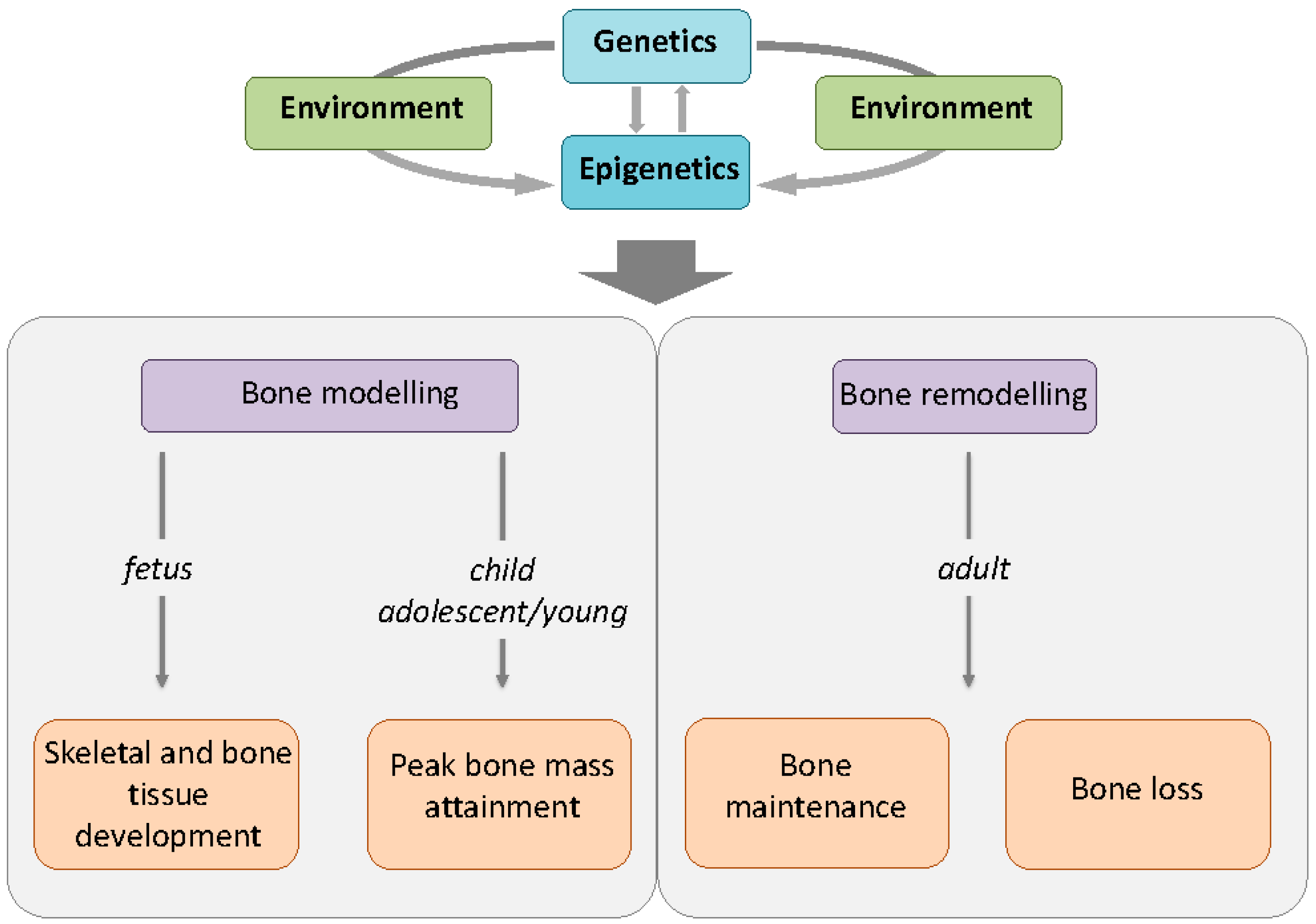
:
1. Introduction
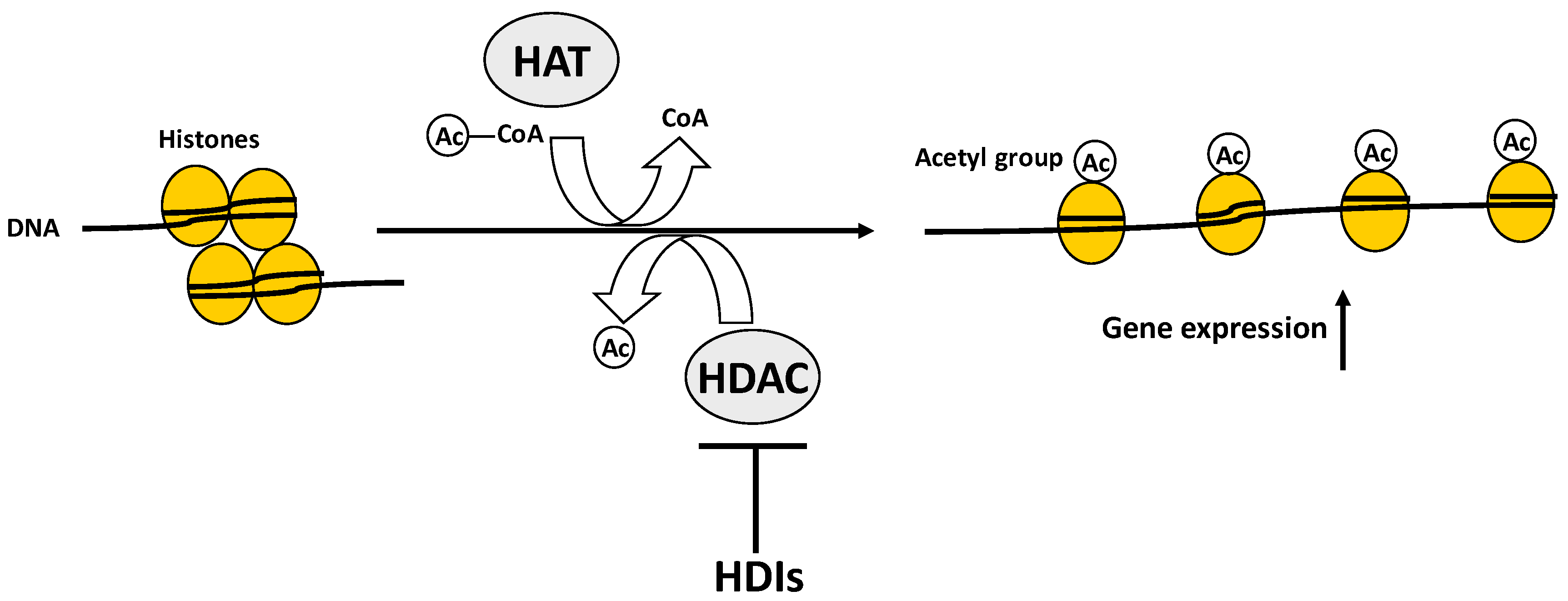
2. Post-Translational Histone Modifications in Bone Biology
3. DNA Methylation in Bone Biology
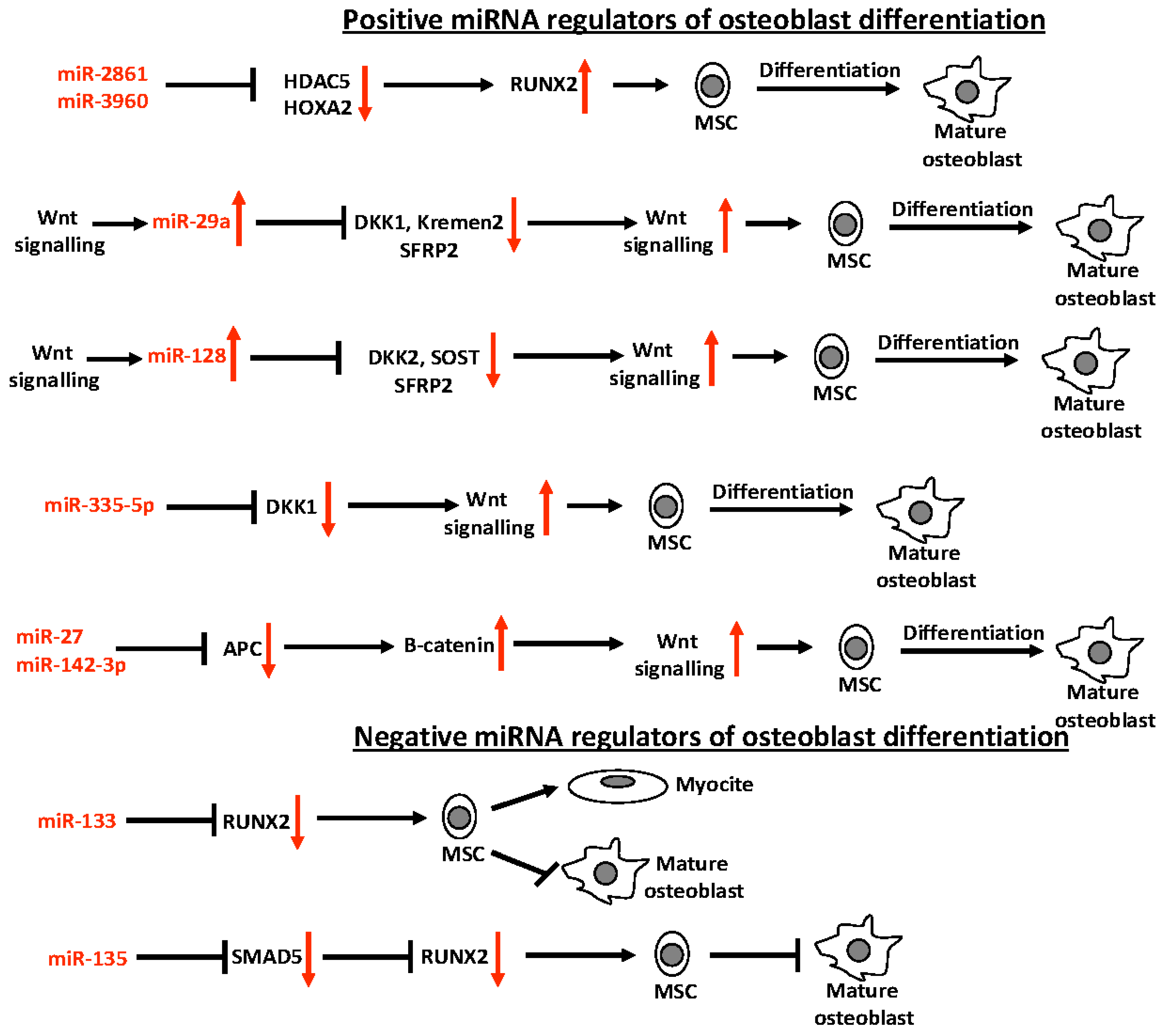
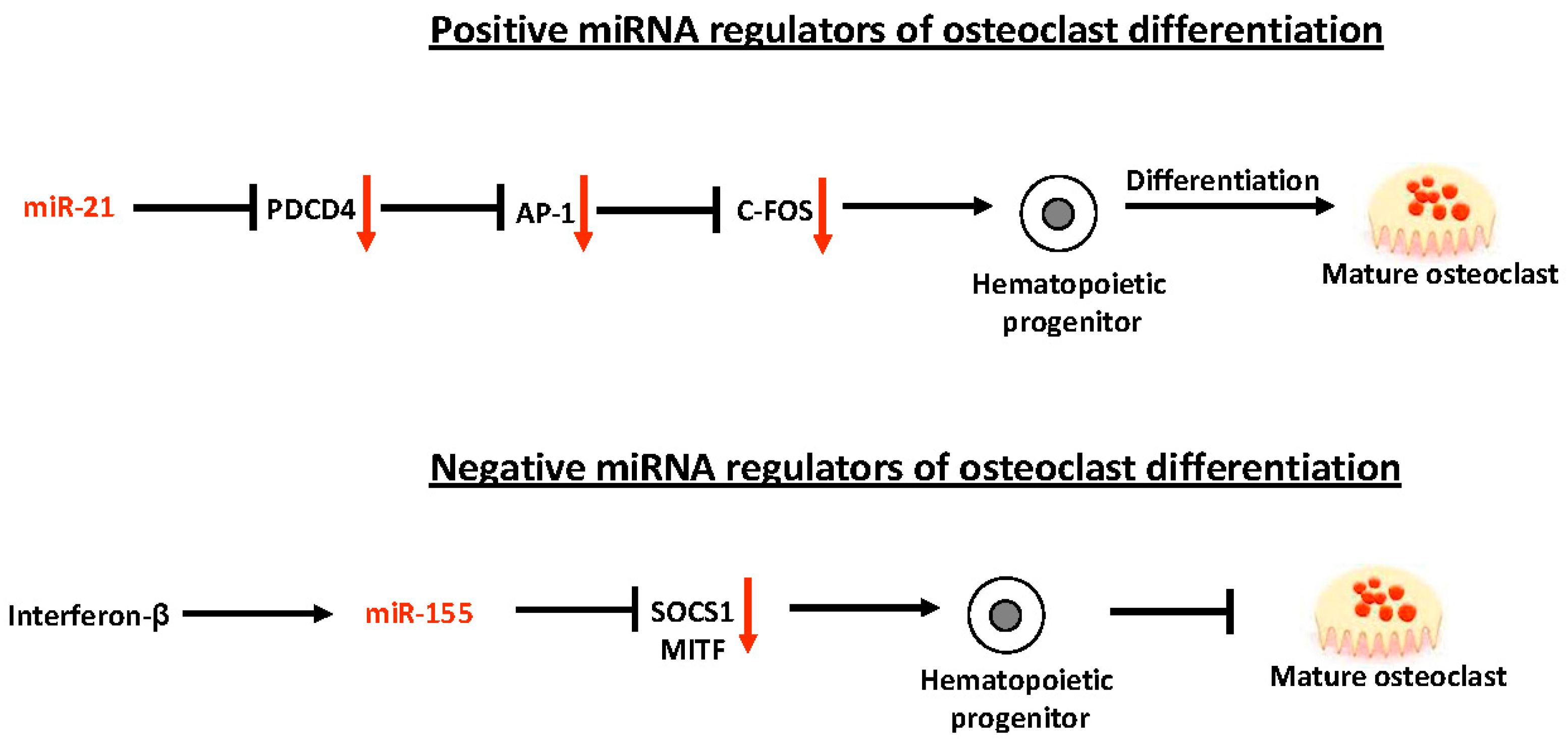
4. miRNAs in Bone Biology
5. Discussion
6. Conclusions and Future Perspectives
Conflicts of Interest
References
- Hsu, Y.H.; Kiel, D.P. Clinical review: Genome-wide association studies of skeletal phenotypes: What we have learned and where we are headed. J. Clin. Endocrinol. Metab. 2012, 97, 1958–1977. [Google Scholar] [CrossRef] [PubMed]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity (Edinburgh) 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Suh, J.H.; Kim, A.Y.; Lee, Y.S.; Park, S.Y.; Kim, J.B. Histone deacetylase 1-mediated histone modification regulates osteoblast differentiation. Mol. Endocrinol. 2006, 20, 2432–2443. [Google Scholar] [CrossRef] [PubMed]
- Dou, C.; Li, N.; Ding, N.; Liu, C.; Yang, X.; Kang, F.; Cao, Z.; Quan, H.; Hou, T.; Xu, J.; Dong, S. HDAC2 regulates FoxO1 during RANKL-induced osteoclastogenesis. Am. J. Physiol. Cell Physiol. 2016, 310, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, T.M.; Kahler, R.A.; Li, X.; Westendorf, J.J. Histone deacetylase 3 interacts with RUNX2 to repress the osteocalcin promoter and regulate osteoblast differentiation. J. Biol. Chem. 2004, 279, 41998–42007. [Google Scholar] [CrossRef] [PubMed]
- Razidlo, D.F.; Whitney, T.J.; Casper, M.E.; McGee-Lawrence, M.E.; Stensgard, B.A.; Li, X.; Secreto, F.J.; Knutson, S.K.; Hiebert, S.W.; Westendorf, J.J. Histone deacetylase 3 depletion in osteo/chondroprogenitor cells decreases bone density and increases marrow fat. PLoS ONE 2010, 5, e11492. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.W.; Hiebert, S.W. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell 2008, 30, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Alliston, T.; Delston, R.; Derynck, R. Repression of RUNX2 function by TGF-β through recruitment of class II histone deacetylases by Smad3. EMBO J. 2005, 24, 2543–2555. [Google Scholar] [CrossRef] [PubMed]
- Hug, B.A. HDAC4: A corepressor controlling bone development. Cell 2004, 119, 448–449. [Google Scholar] [CrossRef] [PubMed]
- Pei, M.; Chen, D.; Li, J.; Wei, L. Histone deacetylase 4 promotes TGF-β1-induced synovium-derived stem cell chondrogenesis but inhibits chondrogenically differentiated stem cell hypertrophy. Differentiation 2009, 78, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Mokalled, M.H.; Montgomery, R.L.; Olson, E.N. Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev. 2009, 23, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Decroos, C.; Bowman, C.M.; Moser, J.A.; Christianson, K.E.; Deardorff, M.A.; Christianson, D.W. Compromised structure and function of HDAC8 mutants identified in Cornelia de Lange Syndrome spectrum disorders. ACS Chem. Biol. 2014, 9, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Harakalova, M.; van den Boogaard, M.J.; Sinke, R.; van Lieshout, S.; van Tuil, M.C.; Duran, K.; Renkens, I.; Terhal, P.A.; de Kovel, C.; Nijman, I.J.; et al. X-exome sequencing identifies a HDAC8 variant in a large pedigree with X-linked intellectual disability, truncal obesity, gynaecomastia, hypogonadism and unusual face. J. Med. Genet. 2012, 49, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xie, H.; Liu, W.; Hu, R.; Huang, B.; Tan, Y.F.; Xu, K.; Sheng, Z.F.; Zhou, H.D.; Wu, X.P.; et al. A novel microRNA targeting HDAC5 regulates osteoblast differentiation in mice and contributes to primary osteoporosis in humans. J. Clin. Investig. 2009, 119, 3666–3677. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.D.; Schroeder, T.M.; Bailey, J.; Gopalakrishnan, R.; Westendorf, J.J. Histone deacetylase 7 associates with RUNX2 and represses its activity during osteoblast maturation in a deacetylation-independent manner. J. Bone Miner. Res. 2008, 23, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Wei, W.; Dechow, P.C.; Wan, Y. HDAC7 inhibits osteoclastogenesis by reversing RANKL-triggered β-catenin switch. Mol. Endocrinol. 2013, 27, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Bradley, E.W.; Carpio, L.R.; van Wijnen, A.J.; McGee-Lawrence, M.E.; Westendorf, J.J. Histone deacetylases in bone development and skeletal disorders. Physiol. Rev. 2015, 95, 1359–1381. [Google Scholar] [CrossRef] [PubMed]
- Backesjo, C.M.; Li, Y.; Lindgren, U.; Haldosen, L.A. Activation of Sirt1 decreases adipocyte formation during osteoblast differentiation of mesenchymal stem cells. J. Bone Miner. Res. 2006, 21, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, T.M.; Westendorf, J.J. Histone deacetylase inhibitors promote osteoblast maturation. J. Bone Miner. Res. 2007, 20, 2254–2263. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Kukita, A.; Kukita, T.; Shobuike, T.; Nakamura, T.; Kohashi, O. Two histone deacetylase inhibitors, trichostatin A and sodium butyrate, suppress differentiation into osteoclasts but not into macrophages. Blood 2003, 101, 3451–3459. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Baek, J.H.; Kim, H.J.; Choi, M.H.; Seo, S.B.; Ryoo, H.M.; Kim, G.S.; Woo, K.M. Trichostatin A-mediated upregulation of p21 (WAF1) contributes to osteoclast apoptosis. Exp. Mol. Med. 2007, 39, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Roy, E.M.; Murphy, T.C.; Nanes, M.S.; Kim, S.; Pike, J.W.; Rubin, J. Regulation of RANKL promoter activity is associated with histone remodeling in murine bone stromal cells. J. Cell. Biochem. 2004, 93, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Senn, S.M.; Kantor, S.; Poulton, I.J.; Morris, M.J.; Sims, N.A.; O’Brien, T.J.; Wark, J.D. Adverse effects of valproate on bone: Defining a model to investigate the pathophysiology. Epilepsia 2010, 51, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Nissen-Meyer, L.S.; Svalheim, S.; Tauboll, E.; Reppe, S.; Lekva, T.; Solberg, L.B.; Melhus, G.; Reinholt, F.P.; Gjerstad, L.; Jemtland, R. Levetiracetam, phenytoin, and valproate act differently on rat bone mass, structure, and metabolism. Epilepsia 2007, 48, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Boluk, A.; Guzelipek, M.; Savli, H.; Temel, I.; Ozişik, H.I.; Kaygusuz, A. The effect of valproate on bone mineral density in adult epileptic patients. Pharmacol. Res. 2004, 50, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.O.; Jacobson, M.P.; Haneef, Z. Homocysteine and bone loss in epilepsy. Seizure 2007, 16, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, P.; Rejnmark, L.; Mosekilde, L. Fracture risk associated with use of antiepileptic drugs. Epilepsia 2004, 45, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Liu, S.; Ma, S.; Zhao, J.; Zhang, W.; Qi, W.; Cao, P.; Wang, Z.; Lei, W. Protective effects of resveratrol on post-menopausal osteoporosis: Regulation of Sirt1-NF-κB signaling pathway. Acta Biochim. Biophys. Sin. 2014, 46, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.; Chen, Q.M.; Hong, C.; Wang, C.Y. Histone methyltransferases and demethylases: Regulators in balancing osteogenic and adipogenic differentiation of mesenchymal stem cells. Int. J. Oral Sci. 2015, 7, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Vrtacnik, P.; Marc, J.; Ostanek, B. Epigenetic mechanisms in bone. Clin. Chem. Lab. Med. 2014, 52, 589–608. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Iwamoto, Y.; Kobayashi, Y.; Katsuoka, F.; Kawaguchi, S.; Tsujita, T.; Nakamura, T.; Kato, S.; Yamamoto, M.; Takayanagi, H.; et al. DNA methyltransferase 3a regulates osteoclast differentiation by coupling to an S-adenosylmethionine-producing metabolic pathway. Nat. Med. 2015, 21, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Sanudo, C.; Fernandez, A.F.; Garcia-Renedo, R.; Fraga, M.F.; Riancho, J.A. Role of DNA methylation in the regulation of the RANKL-OPG system in human bone. Epigenetics 2012, 7, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Reppe, S.; Noer, A.; Grimholt, R.M.; Halldorsson, B.V.; Medina-Gomez, C.; Gautvik, V.T.; Olstad, O.K.; Berg, J.P.; Datta, H.; Estrada, K.; et al. Methylation of bone SOST, its mRNA, and serum sclerostin levels correlate strongly with fracture risk in post-menopausal women. J. Bone Miner. Res. 2015, 30, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Fernandez, A.F.; Sainz, J.; Zarrabeitia, M.T.; Sanudo, C.; Garcia-Renedo, R.; Perez-Nunez, M.I.; Garcia-Ibarbia, C.; Fraga, M.F.; Riancho, J.A. Genome-wide profiling of bone reveals differentially methylated regions in osteoporosis and osteoarthritis. Arthritis Rheum. 2013, 65, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Mahon, P.; Harvey, N.; Crozier, S.; Inskip, H.; Robinson, S.; Arden, N.; Swaminathan, R.; Cooper, C.; Godfrey, K. Low maternal vitamin D status and fetal bone development: Cohort study. J. Bone Miner. Res. 2010, 25, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Oreffo, R.O.; Lashbrooke, B.; Roach, H.I.; Clarke, N.M.; Cooper, C. Maternal protein deficiency affects mesenchymal stem cell activity in the developing offspring. Bone 2003, 33, 100–107. [Google Scholar] [CrossRef]
- Lyllicrop, K.A.; Phillips, E.S.; Torrens, C.; Hanson, M.A.; Jackson, A.A.; Burdge, G.C. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPARα promoter of the offspring. Br. J. Nutr. 2008, 100, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Lyllicrop, K.A.; Slater-Jefferies, J.L.; Hanson, M.A.; Godfrey, K.M.; Jackson, A.A.; Burdge, G.C. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br. J. Nutr. 2007, 97, 1064–1073. [Google Scholar]
- Gaur, T.; Hussain, S.; Mudhasani, R.; Parulkar, I.; Colby, J.L.; Frederick, D.; Kream, B.E.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; et al. Dicer inactivation in osteoprogenitor cells compromises fetal survival and bone formation, while excision in differentiated osteoblasts increases bone mass in the adult mouse. Dev. Biol. 2010, 40, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, F.; Izu, Y.; Hayata, T.; Hemmi, H.; Nakashima, K.; Nakamura, T.; Kato, S.; Miyasaka, N.; Ezura, Y.; Noda, M. Osteoclast-specific Dicer gene deficiency suppresses osteoclastic bone resorption. J. Cell. Biochem. 2010, 109, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, R.L.; Croce, C.M.; Stein, J.L.; Lian, J.B.; van Wijnen, A.J.; Stein, G.S. A program of microRNAs controls osteogenic lineage progression by targeting transcription factor RUNX2. Proc. Natl. Acad. Sci. USA 2011, 108, 9863–9868. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hassan, M.Q.; Volinia, S.; van Wijnen, A.J.; Stein, J.L.; Croce, C.M.; Lian, J.B.; Stein, G.S. A microRNA signature for a BMP2-induced osteoblast lineage commitment program. Proc. Natl. Acad. Sci. USA 2008, 105, 13906–13911. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhao, L.; Xing, L.; Chen, D. MicroRNA-204 regulates RUNX2 protein expression and mesenchymal progenitor cell differentiation. Stem Cells 2010, 28, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kang, I.H.; Lee, J.W.; Jang, W.G.; Koh, J.T. miR-433 mediates ERRγ-suppressed osteoblast differentiation via direct targeting to RUNX2 mRNA in C3H10T1/2 cells. Life Sci. 2013, 92, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Liu, W.; Li, H.; Yang, L.; Chen, C.; Xia, Z.Y.; Guo, L.J.; Xie, H.; Zhou, H.D.; Wu, X.P.; et al. A RUNX2/miR-3960/miR-2861 regulatory feedback loop during mouse osteoblast differentiation. J. Biol. Chem. 2011, 286, 12328–12339. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.Q.; Gordon, J.A.; Beloti, M.M.; Croce, C.M.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. A network connecting RUNX2, SATB2, and the miR-23a~27a~24–2 cluster regulates the osteoblast differentiation program. Proc. Natl. Acad. Sci. USA 2010, 107, 19879–19884. [Google Scholar] [CrossRef] [PubMed]
- Kapinas, K.; Kessler, C.; Ricks, T.; Gronowicz, G.; Delany, A.M. miR-29 modulates Wnt signaling in human osteoblasts through a positive feedback loop. J. Biol. Chem. 2010, 285, 25221–25231. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.Q.; Maeda, Y.; Taipaleenmaki, H.; Zhang, W.; Jafferji, M.; Gordon, J.A.; Li, Z.; Croce, C.M.; van Wijnen, A.J.; Stein, J.L.; et al. miR-218 directs a Wnt signaling circuit to promote differentiation of osteoblasts and osteomimicry of metastatic cancer cells. J. Biol. Chem. 2012, 287, 42084–42092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tu, Q.; Bonewald, L.F.; He, X.; Stein, G.; Lian, J.; Chen, J. Effects of miR-335-5p in modulating osteogenic differentiation by specifically downregulating Wnt antagonist DKK1. J. Bone Miner. Res. 2011, 26, 1953–1963. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xu, Z. miR-27 promotes osteoblast differentiation by modulating Wnt signaling. Biochem. Biophys. Res. Commun. 2010, 402, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Ye, Y.; Zhang, W.; Wang, J.; Chen, A.; Guo, F. miR-142-3p promotes osteoblast differentiation by modulating Wnt signaling. Mol. Med. Rep. 2013, 7, 689–693. [Google Scholar] [PubMed]
- Sugatani, T.; Vacher, J.; Hruska, K.A. A microRNA expression signature of osteoclastogenesis. Blood 2011, 117, 3648–3657. [Google Scholar] [CrossRef] [PubMed]
- Sugatani, T.; Hruska, K.A. Down-regulation of miR-21 biogenesis by estrogen action contributes to osteoclastic apoptosis. J. Cell. Biochem. 2013, 114, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, H.; Chen, J.; Xia, B.; Jin, Y.; Wei, W.; Shen, J.; Huang, Y. Interferon-β-induced miR-155 inhibits osteoclast differentiation by targeting SOCS1 and MITF. FEBS Lett. 2012, 586, 3255–3262. [Google Scholar] [CrossRef] [PubMed]
- Sugatani, T.; Hruska, K.A. MicroRNA-223 is a key factor in osteoclast differentiation. J. Cell. Biochem. 2007, 101, 996–999. [Google Scholar] [CrossRef] [PubMed]
- M'Baya-Moutoula, E.; Louvet, L.; Metzinger-Le Meuth, V.; Massy, Z.A.; Metzinger, L. High inorganic phosphate concentration inhibits osteoclastogenesis by modulating miR-223. Biochim. Biophys. Acta 2015, 1852, 2202–2212. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The microRNA spectrum in 12 body fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Z.; Fu, Q.; Zhang, J. Plasma miRNA levels correlate with sensitivity to bone mineral density in post-menopausal osteoporosis patients. Biomarkers 2014, 19, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, C.; Karpinski, K.; Haug, A.T.; Vester, H.; Schmitt, A.; Bauer, J.S.; van Griensven, M. Five freely circulating miRNAs and bone tissue miRNAs are associated with osteoporotic fractures. J. Bone Miner. Res. 2014, 29, 1718–1728. [Google Scholar] [CrossRef] [PubMed]
- Weilner, S.; Skalicky, S.; Salzer, B.; Keider, V.; Wagner, M.; Hildner, F.; Gabriel, C.; Dovjak, P.; Pietschmann, P.; Grillari-Voglauer, R.; et al. Differentially circulating miRNAs after recent osteoporotic fractures can influence osteogenic differentiation. Bone 2015, 79, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Panach, L.; Mifsut, D.; Tarin, J.J.; Cano, A.; Garcia-Perez, M.A. Serum circulating microRNAs as biomarkers of osteoporotic fracture. Calcif. Tissue Int. 2015, 97, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Szarc vel Szic, K.; Declerck, K.; Vidakovic, M.; Vanden Berghe, W. From inflammaging to healthy aging by dietary lifestyle choices: Is epigenetics the key to personalized nutrition? Clin. Epigenet. 2015, 7, 33–51. [Google Scholar] [CrossRef] [PubMed]
- De Bono, S.; Schoenmakers, I.; Ceesay, M.; Mendy, M.; Laskey, M.A.; Cole, TJ.; Prentice, A. Birth weight predicts bone size in young adulthood at cortical sites in men and trabecular sites in women from The Gambia. Bone 2010, 46, 1316–1321. [Google Scholar] [CrossRef] [PubMed]
- Schlüssel, M.M.; de Castro, J.A.; Kac, G.; da Silva, A.A.; Cardoso, V.C.; Bettiol, H.; Barbieri, M.A. Birth weight and bone mass in young adults from Brazil. Bone 2010, 46, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Dennison, E.M.; Syddall, H.E.; Sayer, A.A.; Gilbody, H.J.; Cooper, C. Birth weight and weight at 1 year are independent determinants of bone mass in the seventh decade: The Hertfordshire cohort study. Pediatr. Res. 2005, 57, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Bocheva, G.; Boyadjieva, N. Epigenetic regulation of fetal bone development and placental transfer of nutrients: Progress for osteoporosis. Interdiscip. Toxicol. 2011, 4, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Wang, K.Y.; Shen, C.K. DNA 5-methylcytosine demethylation activities of the mammalian DNA methyltransferases. J. Biol. Chem. 2013, 288, 9084–9091. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
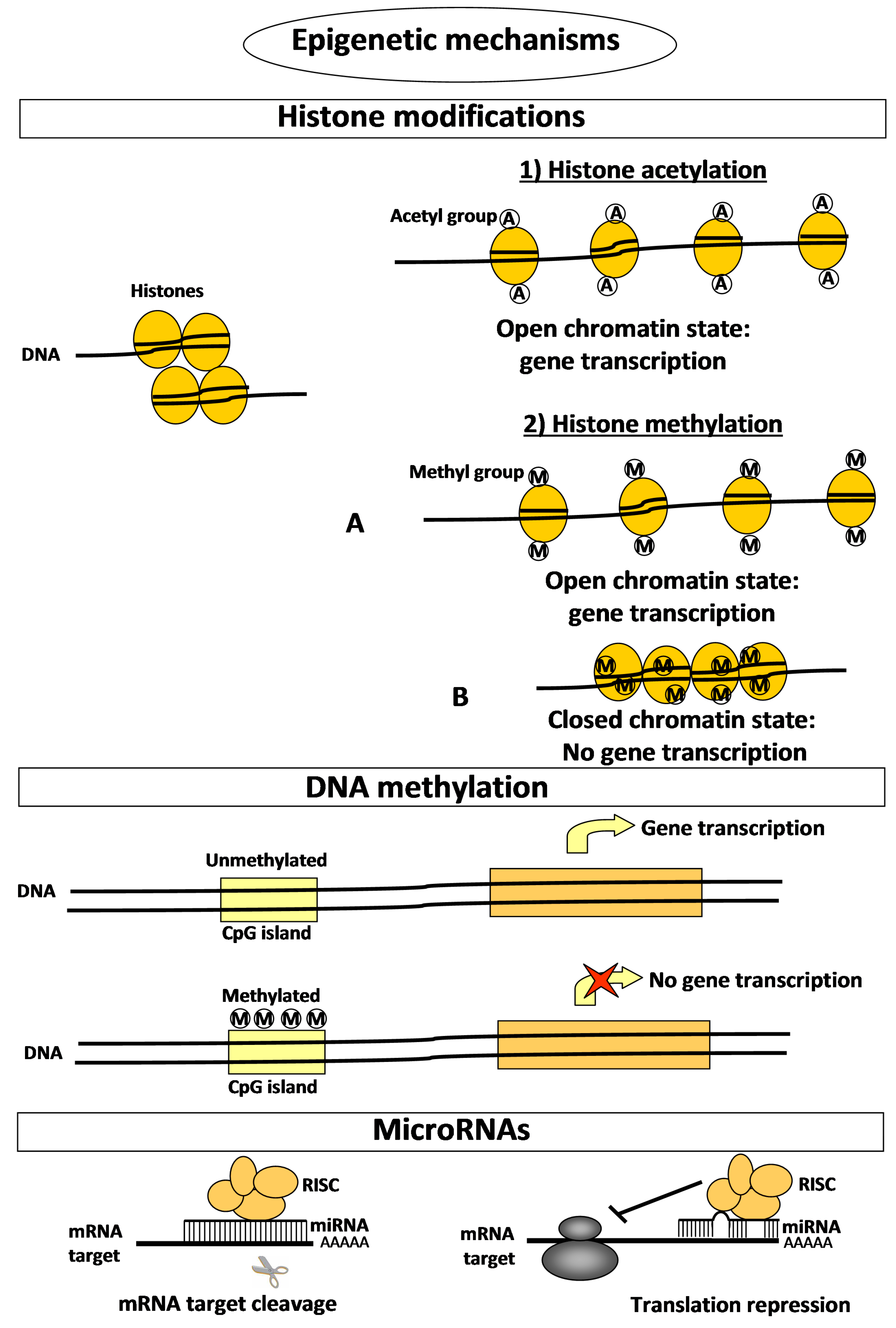
| Epigenetic Process (Post-Translational Histone Modifications) | Molecular Mechanism | Involved Enzymes | Mechanism of Action | Effects on Gene Expression |
|---|---|---|---|---|
| Histone acetylation/deacetylation | The lysine residues at the N-terminal of histone tails are subjected to either addition (acetylation) or removal (deacetylation) of acetyl groups. | (1) Histone acetyltransferases (HATs); | Acetylation removes positive charges from lysine residues and reduces the affinity between histones and DNA, thereby opening the condensed chromatin structure, favouring the access to gene promoters. | Histone acetylation promotes gene expression. Conversely, histone deacetylation prevents gene expression. |
| (2) Histone deacetylases (HDACs) | ||||
| Histone methylation/demethylation | Histone methylation occurs on different lysine residues, with the potential addition of one, two, or three methyl groups. | (1) Histone lysine methyltransferases (KMTs); | The effect of histone methylation on chromatin state is dependent not only on the specific lysine residue modified, but also on its degree of methylation. | Histone methylation at H3K4, H3K36, or H3K79 has been associated with gene transcription activation. |
| (2) Histone lysine demethylases (KDMs) | Histone methylation at H3K9, H3K20, or H4K27 is implicated in gene expression inactivation or silencing. | |||
| DNA methylation | Addition of a methyl group at the 5′ position of the cytosine ring within CpG islands of gene promoters. | (1) DNA methyltransferases (DNMT3A and DNMT3B); | Methylated gene promoters are not accessible to transcription factors. | DNA methylation is strongly associated with gene transcription silencing. |
| (2) DNA maintenance methyltransferase (DNMT1) | ||||
| MicroRNAs (miRNAs) | miRNAs selectively bind to the 3’ non coding region (3’UTR) of specific target mRNAs, through base-pairing. | None | Binding of a miRNA on the 3’UTR of the target mRNA blocks protein synthesis by two distinct post-transcriptional mechanisms: mRNA cleavage or translational repression. | miRNAs negatively regulate the expression of target genes, at post-transcriptional level, by blocking the translation of their proteins. |
| HDAC | Class | Affected Protein Expression | Effects on Bone Biology | Reference |
|---|---|---|---|---|
| HDAC1 | I | RUNX2 (down-regulation) | Suppression of osteoblast differentiation | [6] |
| HDAC2 | I | FoxO1 (down-regulation) | Promotion of RANKL-induced osteoclastogenesis | [13] |
| HDAC3 | I | RUNX2 (down-regulation) | Maintenance of bone mass during development and aging | [3] |
| HDAC4 | II | RUNX2 (down-regulation) | Suppression of endochondral ossification | [8,9] |
| HDAC5 | II | RUNX2 (down-regulation) | Suppression of osteoblast differentiation | [8] |
| HDAC7 | II | RUNX2 (down-regulation) | Regulation of endochondral ossification | [15] |
| HDAC8 | I | Homeobox transcription factors Otx2 (up-regulation) and Lhx1 (up-regulation) | Regulation of intramembranous ossification | [11] |
| HDAC9 | II | RANKL (down-regulation) | Suppression of osteoclastogenesis | [17] |
| Sirt1 | III | NA | Promotion of endochondral ossification, and of osteoblast differentiation of mesenchymal stem cells | [17,18] |
| Sirt6 | III | NA | Promotion of endochondral ossification | [17] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marini, F.; Cianferotti, L.; Brandi, M.L. Epigenetic Mechanisms in Bone Biology and Osteoporosis: Can They Drive Therapeutic Choices? Int. J. Mol. Sci. 2016, 17, 1329. https://doi.org/10.3390/ijms17081329
Marini F, Cianferotti L, Brandi ML. Epigenetic Mechanisms in Bone Biology and Osteoporosis: Can They Drive Therapeutic Choices? International Journal of Molecular Sciences. 2016; 17(8):1329. https://doi.org/10.3390/ijms17081329
Chicago/Turabian StyleMarini, Francesca, Luisella Cianferotti, and Maria Luisa Brandi. 2016. "Epigenetic Mechanisms in Bone Biology and Osteoporosis: Can They Drive Therapeutic Choices?" International Journal of Molecular Sciences 17, no. 8: 1329. https://doi.org/10.3390/ijms17081329
APA StyleMarini, F., Cianferotti, L., & Brandi, M. L. (2016). Epigenetic Mechanisms in Bone Biology and Osteoporosis: Can They Drive Therapeutic Choices? International Journal of Molecular Sciences, 17(8), 1329. https://doi.org/10.3390/ijms17081329