
Pharmacogenomics in Pediatric Oncology: Review of Gene—Drug Associations for Clinical Use †

Abstract
:
1. Introduction
2. The Role of Ontogeny in Pharmacogenomics
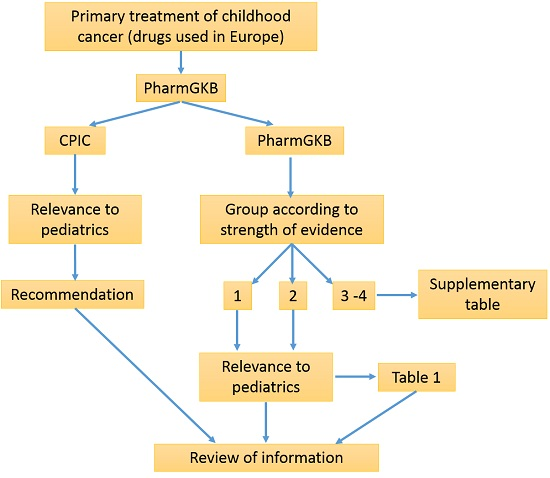
3. Methods
4. Results
4.1. Drugs with Strong Pharmacogenetics Evidence
4.1.1. Thiopurines/Thiopurine S-Methyltransferases (TPMT) Pair
4.1.2. Thipurines/Nudix Hydrolase 15 (NUDT15) Pair
4.1.3. Cisplatin/Xeroderma Pigmentosum, Complementation Group C (XPC) Pair
4.2. Drugs with Moderate Pharmacogenetics Evidence
4.2.1. Cisplatin and Carboplatin
4.2.2. Methotrexate
4.2.3. Cyclophosphamide
4.2.4. Irinotecan
4.2.5. Vincristine
5. Conclusions
6. Future Directions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pritchard-Jones, K.; Dixon-Woods, M.; Naafs-Wilstra, M.; Valsecchi, M.G. Improving recruitment to clinical trials for cancer in childhood. Lancet Oncol. 2008, 9, 392–399. [Google Scholar] [CrossRef]
- Mitchell, A.A.; Lacouture, P.G.; Sheehan, J.E.; Kauffman, R.E.; Shapiro, S. Adverse drug reactions in children leading to hospital admission. Pediatrics 1988, 82, 24–29. [Google Scholar] [PubMed]
- MacNeil, M.; Eisenhauer, E.A. High-dose chemotherapy: Is it standard management for any common solid tumor? Ann. Oncol. 1999, 10, 1145–1161. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W. Pharmacogenetics and pharmacogenomics: Why is this relevant to the clinical geneticist? Clin. Genet. 1999, 56, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.; de Leonibus, C.; Hanson, D.; Whatmore, A.; Murray, P.; Donn, R.; Meyer, S.; Chatelain, P.; Clayton, P. Pediatric perspective on pharmacogenomics. Pharmacogenomics 2013, 14, 1889–1905. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shalom, D.; Rose, K. Pediatric Formulations: A Roadmap; AAPS Press/Springer: New York, NY, USA, 2014; Volume 11. [Google Scholar]
- Kearns, G.L.; Abdel-Rahman, S.M.; Alander, S.W.; Blowey, D.L.; Leeder, J.S.; Kauffman, R.E. Developmental pharmacology—Drug disposition, action, and therapy in infants and children. N. Engl. J. Med. 2003, 349, 1157–1167. [Google Scholar] [PubMed]
- Leeder, J.S.; Kearns, G.L. Pharmacogenetics in pediatrics. Implications for practice. Pediatr. Clin. N. Am. 1997, 44, 55–77. [Google Scholar] [CrossRef]
- De Wildt, S.N.; Kearns, G.L.; Leeder, J.S.; van den Anker, J.N. Cytochrome P450 3A: Ontogeny and drug disposition. Clin. Pharmacokinet. 1999, 37, 485–505. [Google Scholar] [CrossRef] [PubMed]
- Treluyer, J.M.; Jacqz-Aigrain, E.; Alvarez, F.; Cresteil, T. Expression of CYP2D6 in developing human liver. Eur. J. Biochem. 1991, 202, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, K.L.; Aleksunes, L.M.; Brandys, B.; Giacoia, G.P.; Knipp, G.; Lukacova, V.; Meibohm, B.; Nigam, S.K.; Rieder, M.; de Wildt, S.N.; et al. Human Ontogeny of Drug Transporters: Review and Recommendations of the Pediatric Transporter Working Group. Clin. Pharmacol. Ther. 2015, 98, 266–287. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.; Hanson, D.; Whatmore, A.; Destenaves, B.; Chatelain, P.; Clayton, P. Human growth is associated with distinct patterns of gene expression in evolutionarily conserved networks. BMC Genom. 2013, 14, 547. [Google Scholar] [CrossRef] [PubMed]
- Finkielstain, G.P.; Forcinito, P.; Lui, J.C.; Barnes, K.M.; Marino, R.; Makaroun, S.; Nguyen, V.; Lazarus, J.E.; Nilsson, O.; Baron, J. An extensive genetic program occurring during postnatal growth in multiple tissues. Endocrinology 2009, 150, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Knight, K.R.; Kraemer, D.F.; Neuwelt, E.A. Ototoxicity in children receiving platinum chemotherapy: Underestimating a commonly occurring toxicity that may influence academic and social development. J. Clin. Oncol. 2005, 23, 8588–8596. [Google Scholar] [CrossRef] [PubMed]
- Kushner, B.H.; Budnick, A.; Kramer, K.; Modak, S.; Cheung, N.K. Ototoxicity from high-dose use of platinum compounds in patients with neuroblastoma. Cancer 2006, 107, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Bleyer, W.A.; Fallavollita, J.; Robison, L.; Balsom, W.; Meadows, A.; Heyn, R.; Sitarz, A.; Ortega, J.; Miller, D.; Constine, L.; et al. Influence of age, sex, and concurrent intrathecal methotrexate therapy on intellectual function after cranial irradiation during childhood: A report from the Children’s Cancer Study Group. Pediatr. Hematol. Oncol. 1990, 7, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Lazaryan, M.; Shasha-Zigelman, C.; Dagan, Z.; Berkovitch, M. Codeine should not be prescribed for breastfeeding mothers or children under the age of 12. Acta Paediatr. 2015, 104, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Uppugunduri, C.R.; Ansari, M. Commentary: A myriad aberrations on information of ontogeny of drug metabolizing enzymes in the pediatric population: An obstacle for personalizing drug therapy in the pediatric population. Drug Metab. Lett. 2016, 10, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.C.; Doll, D.C.; Freter, C.E. Chemotherapy Source Book, 5th ed.; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012. [Google Scholar]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Aghdaie, M.H.; Azarpira, N.; Geramizadeh, B.; Sagheb, M.; Darai, M.; Rahsaz, M.; Malekhoseini, S.A. Thiopurine S-methyltransferase polymorphism in Iranian kidney transplant recipients. Exp. Clin. Transplant. 2011, 9, 241–246. [Google Scholar] [PubMed]
- Alves, S.; Amorim, A.; Ferreira, F.; Prata, M.J. Influence of the variable number of tandem repeats located in the promoter region of the thiopurine methyltransferase gene on enzymatic activity. Clin. Pharmacol. Ther. 2001, 70, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Alves, S.; Prata, M.J.; Ferreira, F.; Amorim, A. Thiopurine methyltransferase pharmacogenetics: Alternative molecular diagnosis and preliminary data from Northern Portugal. Pharmacogenetics 1999, 9, 257–261. [Google Scholar] [PubMed]
- Ansari, A.; Arenas, M.; Greenfield, S.M.; Morris, D.; Lindsay, J.; Gilshenan, K.; Smith, M.; Lewis, C.; Marinaki, A.; Duley, J.; et al. Prospective evaluation of the pharmacogenetics of azathioprine in the treatment of inflammatory bowel disease. Aliment. Pharmacol. Ther. 2008, 28, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.; Hassan, C.; Duley, J.; Marinaki, A.; Shobowale-Bakre, E.M.; Seed, P.; Meenan, J.; Yim, A.; Sanderson, J. Thiopurine methyltransferase activity and the use of azathioprine in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2002, 16, 1743–1750. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.; Andoh, A.; Tanaka, A.; Tsujikawa, T.; Sasaki, M.; Saito, Y.; Fujiyama, Y. Analysis of thiopurine S-methyltransferase genotypes in Japanese patients with inflammatory bowel disease. Intern. Med. 2008, 47, 1645–1648. [Google Scholar] [CrossRef] [PubMed]
- Black, A.J.; McLeod, H.L.; Capell, H.A.; Powrie, R.H.; Matowe, L.K.; Pritchard, S.C.; Collie-Duguid, E.S.; Reid, D.M. Thiopurine methyltransferase genotype predicts therapy-limiting severe toxicity from azathioprine. Ann. Intern. Med. 1998, 129, 716–718. [Google Scholar] [CrossRef] [PubMed]
- Breen, D.P.; Marinaki, A.M.; Arenas, M.; Hayes, P.C. Pharmacogenetic association with adverse drug reactions to azathioprine immunosuppressive therapy following liver transplantation. Liver Transpl. 2005, 11, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Budhiraja, P.; Popovtzer, M. Azathioprine-related myelosuppression in a patient homozygous for TPMT*3A. Nat. Rev. Nephrol. 2011, 7, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.T.; Allan, R.N. Mistaken identity: Misclassification of TPMT phenotype following blood transfusion. Eur. J. Gastroenterol. Hepatol. 2003, 15, 1245–1247. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska, M.; Kurzawski, M.; Drozdzik, M.; Mazik, M.; Oko, A.; Czekalski, S. Thiopurine S-methyltransferase phenotype-genotype correlation in hemodialyzed patients. Pharmacol. Rep. 2006, 58, 973–978. [Google Scholar] [PubMed]
- Corominas, H.; Domenech, M.; Laiz, A.; Gich, I.; Geli, C.; Diaz, C.; de Cuevillas, F.; Moreno, M.; Vazquez, G.; Baiget, M. Is thiopurine methyltransferase genetic polymorphism a major factor for withdrawal of azathioprine in rheumatoid arthritis patients? Rheumatology (Oxford) 2003, 42, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.W.; Zheng, Q.; Zhu, M.M.; Tong, J.L.; Ran, Z.H. Thiopurine S-methyltransferase polymorphisms and thiopurine toxicity in treatment of inflammatory bowel disease. World J. Gastroenterol. 2010, 16, 3187–3195. [Google Scholar] [CrossRef] [PubMed]
- El-Sedfy, A.; Chamberlain, R.S. Surgeons and their tools: A history of surgical instruments and their innovators—Part I: Place the scissors on the Mayo stand. Am. Surg. 2014, 80, 1089–1092. [Google Scholar] [PubMed]
- Fabre, M.A.; Jones, D.C.; Bunce, M.; Morris, P.J.; Friend, P.J.; Welsh, K.I.; Marshall, S.E. The impact of thiopurine S-methyltransferase polymorphisms on azathioprine dose 1 year after renal transplantation. Transpl. Int. 2004, 17, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Formea, C.M.; Myers-Huentelman, H.; Wu, R.; Crabtree, J.; Fujita, S.; Hemming, A.; Reed, A.; Howard, R.; Karlix, J.L. Thiopurine S-methyltransferase genotype predicts azathioprine-induced myelotoxicity in kidney transplant recipients. Am. J. Transplant. 2004, 4, 1810–1817. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, S.J.; Gearry, R.B.; Begg, E.J.; Zhang, M.; Barclay, M.L. Thiopurine dose in intermediate and normal metabolizers of thiopurine methyltransferase may differ three-fold. Clin. Gastroenterol. Hepatol. 2008, 6, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Gearry, R.B.; Barclay, M.L.; Roberts, R.L.; Harraway, J.; Zhang, M.; Pike, L.S.; George, P.M.; Florkowski, C.M. Thiopurine methyltransferase and 6-thioguanine nucleotide measurement: Early experience of use in clinical practice. Intern. Med. J. 2005, 35, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Haglund, S.; Lindqvist, M.; Almer, S.; Peterson, C.; Taipalensuu, J. Pyrosequencing of TPMT alleles in a general Swedish population and in patients with inflammatory bowel disease. Clin. Chem. 2004, 50, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, J.M.; Lambson, E.M.; Little, F.; Owen, E.P. Thiopurine methyltransferase (TPMT) heterozygosity and enzyme activity as predictive tests for the development of azathioprine-related adverse events. J. Neurol. Sci. 2005, 231, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Higgs, J.E.; Payne, K.; Roberts, C.; Newman, W.G. Are patients with intermediate TPMT activity at increased risk of myelosuppression when taking thiopurine medications? Pharmacogenomics 2010, 11, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Hindorf, U.; Lindqvist, M.; Peterson, C.; Soderkvist, P.; Strom, M.; Hjortswang, H.; Pousette, A.; Almer, S. Pharmacogenetics during standardised initiation of thiopurine treatment in inflammatory bowel disease. Gut 2006, 55, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Hindorf, U.; Lyrenas, E.; Nilsson, A.; Schmiegelow, K. Monitoring of long-term thiopurine therapy among adults with inflammatory bowel disease. Scand. J. Gastroenterol. 2004, 39, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Hon, Y.Y.; Fessing, M.Y.; Pui, C.H.; Relling, M.V.; Krynetski, E.Y.; Evans, W.E. Polymorphism of the thiopurine S-methyltransferase gene in African-Americans. Hum. Mol. Genet. 1999, 8, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Indjova, D.; Atanasova, S.; Shipkova, M.; Armstrong, V.W.; Oellerich, M.; Svinarov, D. Phenotypic and genotypic analysis of thiopurine S-methyltransferase polymorphism in the bulgarian population. Ther. Drug Monit. 2003, 25, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.B.; Cho, D.Y.; Kang, C.; Bae, S.C. Thiopurine S-methyltransferase polymorphisms and the relationship between the mutant alleles and the adverse effects in systemic lupus erythematosus patients taking azathioprine. Clin. Exp. Rheumatol. 2005, 23, 873–876. [Google Scholar] [PubMed]
- Kaskas, B.A.; Louis, E.; Hindorf, U.; Schaeffeler, E.; Deflandre, J.; Graepler, F.; Schmiegelow, K.; Gregor, M.; Zanger, U.M.; Eichelbaum, M.; et al. Safe treatment of thiopurine S-methyltransferase deficient Crohn’s disease patients with azathioprine. Gut 2003, 52, 140–142. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Cheon, J.H.; Hong, S.S.; Eun, C.S.; Byeon, J.S.; Hong, S.Y.; Kim, B.Y.; Kwon, S.H.; Kim, S.W.; Han, D.S.; et al. Influences of thiopurine methyltransferase genotype and activity on thiopurine-induced leukopenia in Korean patients with inflammatory bowel disease: A retrospective cohort study. J. Clin. Gastroenterol. 2010, 44, e242–e248. [Google Scholar] [CrossRef] [PubMed]
- Krynetski, E.Y.; Schuetz, J.D.; Galpin, A.J.; Pui, C.H.; Relling, M.V.; Evans, W.E. A single point mutation leading to loss of catalytic activity in human thiopurine S-methyltransferase. Proc. Natl. Acad. Sci. USA 1995, 92, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Kurzawski, M.; Dziewanowski, K.; Gawronska-Szklarz, B.; Domanski, L.; Drozdzik, M. The impact of thiopurine S-methyltransferase polymorphism on azathioprine-induced myelotoxicity in renal transplant recipients. Ther. Drug Monit. 2005, 27, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Landy, J.; Bhuva, N.; Marinaki, A.; Mawdsley, J. Novel thiopurine methyltransferase variant TPMT*28 results in a misdiagnosis of TPMT deficiency. Inflamm. Bowel Dis. 2011, 17, 1441–1442. [Google Scholar] [CrossRef] [PubMed]
- Larovere, L.E.; de Kremer, R.D.; Lambooy, L.H.; De Abreu, R.A. Genetic polymorphism of thiopurine S-methyltransferase in Argentina. Ann. Clin. Biochem. 2003, 40, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, M.; Haglund, S.; Almer, S.; Peterson, C.; Taipalensu, J.; Hertervig, E.; Lyrenas, E.; Soderkvist, P. Identification of two novel sequence variants affecting thiopurine methyltransferase enzyme activity. Pharmacogenetics 2004, 14, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, M.; Skoglund, K.; Karlgren, A.; Soderkvist, P.; Peterson, C.; Kidhall, I.; Almer, S. Explaining TPMT genotype/phenotype discrepancy by haplotyping of TPMT*3A and identification of a novel sequence variant, TPMT*23. Pharmacogenet. Genom. 2007, 17, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Loennechen, T.; Yates, C.R.; Fessing, M.Y.; Relling, M.V.; Krynetski, E.Y.; Evans, W.E. Isolation of a human thiopurine S-methyltransferase (TPMT) complementary DNA with a single nucleotide transition A719G (TPMT*3C) and its association with loss of TPMT protein and catalytic activity in humans. Clin. Pharmacol. Ther. 1998, 64, 46–51. [Google Scholar] [CrossRef]
- Milek, M.; Murn, J.; Jaksic, Z.; Lukac Bajalo, J.; Jazbec, J.; Mlinaric Rascan, I. Thiopurine S-methyltransferase pharmacogenetics: Genotype to phenotype correlation in the Slovenian population. Pharmacology 2006, 77, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Moloney, F.J.; Dicker, P.; Conlon, P.J.; Shields, D.C.; Murphy, G.M. The frequency and significance of thiopurine S-methyltransferase gene polymorphisms in azathioprine-treated renal transplant recipients. Br. J. Dermatol. 2006, 154, 1199–1200. [Google Scholar] [CrossRef] [PubMed]
- Newman, W.G.; Payne, K.; Tricker, K.; Roberts, S.A.; Fargher, E.; Pushpakom, S.; Alder, J.E.; Sidgwick, G.P.; Payne, D.; Elliott, R.A.; et al. A pragmatic randomized controlled trial of thiopurine methyltransferase genotyping prior to azathioprine treatment: The TARGET study. Pharmacogenomics 2011, 12, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Ogungbenro, K.; Aarons, L.; Cresim; Epi, C.P.G. Physiologically based pharmacokinetic model for 6-mercpatopurine: Exploring the role of genetic polymorphism in TPMT enzyme activity. Br. J. Clin. Pharmacol. 2015, 80, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Nakamura, K.; Kodama, T.; Ueki, K.; Tsukada, Y.; Maezawa, A.; Tsukamoto, N.; Nojima, Y.; Ishizaki, T.; Horiuchi, R.; et al. Thiopurine methyltransferase genotype and phenotype status in Japanese patients with systemic lupus erythematosus. Biol. Pharm. Bull. 2005, 28, 2117–2119. [Google Scholar] [CrossRef] [PubMed]
- Otterness, D.; Szumlanski, C.; Lennard, L.; Klemetsdal, B.; Aarbakke, J.; Park-Hah, J.O.; Iven, H.; Schmiegelow, K.; Branum, E.; O’Brien, J.; et al. Human thiopurine methyltransferase pharmacogenetics: Gene sequence polymorphisms. Clin. Pharmacol. Ther. 1997, 62, 60–73. [Google Scholar] [CrossRef]
- Otterness, D.M.; Szumlanski, C.L.; Wood, T.C.; Weinshilboum, R.M. Human thiopurine methyltransferase pharmacogenetics. Kindred with a terminal exon splice junction mutation that results in loss of activity. J. Clin. Investig. 1998, 101, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Reis, M.; Santoro, A.; Suarez-Kurtz, G. Thiopurine methyltransferase phenotypes and genotypes in Brazilians. Pharmacogenetics 2003, 13, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.M.; Bianchi, M.; Guarnieri, C.; Barale, R.; Pacifici, G.M. Genotype-phenotype correlation for thiopurine S-methyltransferase in healthy Italian subjects. Eur. J. Clin. Pharmacol. 2001, 57, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Salavaggione, O.E.; Wang, L.; Wiepert, M.; Yee, V.C.; Weinshilboum, R.M. Thiopurine S-methyltransferase pharmacogenetics: Variant allele functional and comparative genomics. Pharmacogenet. Genom. 2005, 15, 801–815. [Google Scholar] [CrossRef]
- Samochatova, E.V.; Chupova, N.V.; Rudneva, A.; Makarova, O.; Nasedkina, T.V.; Fedorova, O.E.; Glotov, A.S.; Kozhekbaeva, Z.; Maiorova, O.A.; Roumyantsev, A.G.; et al. TPMT genetic variations in populations of the Russian Federation. Pediatr. Blood Cancer 2009, 52, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Schaeffeler, E.; Fischer, C.; Brockmeier, D.; Wernet, D.; Moerike, K.; Eichelbaum, M.; Zanger, U.M.; Schwab, M. Comprehensive analysis of thiopurine S-methyltransferase phenotype-genotype correlation in a large population of German-Caucasians and identification of novel TPMT variants. Pharmacogenetics 2004, 14, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Schaffeler, E.; Marx, C.; Fischer, C.; Lang, T.; Behrens, C.; Gregor, M.; Eichelbaum, M.; Zanger, U.M.; Kaskas, B.A. Azathioprine therapy and adverse drug reactions in patients with inflammatory bowel disease: Impact of thiopurine S-methyltransferase polymorphism. Pharmacogenetics 2002, 12, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Sebbag, L.; Boucher, P.; Davelu, P.; Boissonnat, P.; Champsaur, G.; Ninet, J.; Dureau, G.; Obadia, J.F.; Vallon, J.J.; Delaye, J. Thiopurine S-methyltransferase gene polymorphism is predictive of azathioprine-induced myelosuppression in heart transplant recipients. Transplantation 2000, 69, 1524–1527. [Google Scholar] [CrossRef] [PubMed]
- Serpe, L.; Calvo, P.L.; Muntoni, E.; D'Antico, S.; Giaccone, M.; Avagnina, A.; Baldi, M.; Barbera, C.; Curti, F.; Pera, A.; et al. Thiopurine S-methyltransferase pharmacogenetics in a large-scale healthy Italian-Caucasian population: Differences in enzyme activity. Pharmacogenomics 2009, 10, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- Song, D.K.; Zhao, J.; Zhang, L.R. TPMT genotype and its clinical implication in renal transplant recipients with azathioprine treatment. J. Clin. Pharm. Ther. 2006, 31, 627–635. [Google Scholar] [CrossRef] [PubMed]
- De la Moureyre, C.S.V.; Debuysere, H.; Sabbagh, N.; Marez, D.; Vinner, E.; Chevalier, E.D.; Lo Guidice, J.M.; Broly, F. Detection of known and new mutations in the thiopurine S-methyltransferase gene by single-strand conformation polymorphism analysis. Hum. Mutat. 1998, 12, 177–185. [Google Scholar] [CrossRef]
- Tai, H.L.; Fessing, M.Y.; Bonten, E.J.; Yanishevsky, Y.; d'Azzo, A.; Krynetski, E.Y.; Evans, W.E. Enhanced proteasomal degradation of mutant human thiopurine S-methyltransferase (TPMT) in mammalian cells: Mechanism for TPMT protein deficiency inherited by TPMT*2, TPMT*3A, TPMT*3B or TPMT*3C. Pharmacogenetics 1999, 9, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.L.; Krynetski, E.Y.; Schuetz, E.G.; Yanishevski, Y.; Evans, W.E. Enhanced proteolysis of thiopurine S-methyltransferase (TPMT) encoded by mutant alleles in humans (TPMT*3A, TPMT*2): Mechanisms for the genetic polymorphism of TPMT activity. Proc. Natl. Acad. Sci. USA 1997, 94, 6444–6449. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.L.; Krynetski, E.Y.; Yates, C.R.; Loennechen, T.; Fessing, M.Y.; Krynetskaia, N.F.; Evans, W.E. Thiopurine S-methyltransferase deficiency: Two nucleotide transitions define the most prevalent mutant allele associated with loss of catalytic activity in Caucasians. Am. J. Hum. Genet. 1996, 58, 694–702. [Google Scholar] [PubMed]
- Taja-Chayeb, L.; Vidal-Millan, S.; Gutierrez, O.; Ostrosky-Wegman, P.; Duenas-Gonzalez, A.; Candelaria, M. Thiopurine S-methyltransferase gene (TMPT) polymorphisms in a Mexican population of healthy individuals and leukemic patients. Med. Oncol. 2008, 25, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Takatsu, N.; Matsui, T.; Murakami, Y.; Ishihara, H.; Hisabe, T.; Nagahama, T.; Maki, S.; Beppu, T.; Takaki, Y.; Hirai, F.; et al. Adverse reactions to azathioprine cannot be predicted by thiopurine S-methyltransferase genotype in Japanese patients with inflammatory bowel disease. J. Gastroenterol. Hepatol. 2009, 24, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Tamm, R.; Oselin, K.; Kallassalu, K.; Magi, R.; Anier, K.; Remm, M.; Metspalu, A. Thiopurine S-methyltransferase (TPMT) pharmacogenetics: Three new mutations and haplotype analysis in the Estonian population. Clin. Chem. Lab. Med. 2008, 46, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Tamori, A.; Shinzaki, M.; Kosaka, S.; Hayashi, T.; Iwai, S.; Enomoto, M.; Habu, D.; Sakaguchi, H.; Kawada, N.; Hino, M.; et al. Thiopurine S-methyltransferase gene polymorphism in Japanese patients with autoimmune liver diseases. Liver Int. 2007, 27, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Ujiie, S.; Sasaki, T.; Mizugaki, M.; Ishikawa, M.; Hiratsuka, M. Functional characterization of 23 allelic variants of thiopurine S-methyltransferase gene (TPMT*2–*24). Pharmacogenet. Genom. 2008, 18, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Vannaprasaht, S.; Angsuthum, S.; Avihingsanon, Y.; Sirivongs, D.; Pongskul, C.; Makarawate, P.; Praditpornsilpa, K.; Tassaneeyakul, W.; Tassaneeyakul, W. Impact of the heterozygous TPMT*1/*3C genotype on azathioprine-induced myelosuppression in kidney transplant recipients in Thailand. Clin. Ther. 2009, 31, 1524–1533. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.W.; Gaffney, D.; Shapiro, D.; Spooner, R.J.; Marinaki, A.M.; Sanderson, J.D.; Mills, P.R. Assessment of thiopurine methyltransferase enzyme activity is superior to genotype in predicting myelosuppression following azathioprine therapy in patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 2007, 25, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.W.; Xiong, H.; Wu, X.C.; Li, Q.; Xiong, L.; Yu, A.R. Relationships between thiopurine S-methyltransferase polymorphism and azathioprine-related adverse drug reactions in Chinese renal transplant recipients. Eur. J. Clin. Pharmacol. 2009, 65, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Yates, C.R.; Krynetski, E.Y.; Loennechen, T.; Fessing, M.Y.; Tai, H.L.; Pui, C.H.; Relling, M.V.; Evans, W.E. Molecular diagnosis of thiopurine S-methyltransferase deficiency: Genetic basis for azathioprine and mercaptopurine intolerance. Ann. Intern. Med. 1997, 126, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Zelinkova, Z.; Derijks, L.J.; Stokkers, P.C.; Vogels, E.W.; van Kampen, A.H.; Curvers, W.L.; Cohn, D.; van Deventer, S.J.; Hommes, D.W. Inosine triphosphate pyrophosphatase and thiopurine S-methyltransferase genotypes relationship to azathioprine-induced myelosuppression. Clin. Gastroenterol. Hepatol. 2006, 4, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.R.; Song, D.K.; Zhang, W.; Zhao, J.; Jia, L.J.; Xing, D.L. Efficient screening method of the thiopurine methyltransferase polymorphisms for patients considering taking thiopurine drugs in a Chinese Han population in Henan Province (central China). Clin. Chim. Acta 2007, 376, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Adam de Beaumais, T.; Fakhoury, M.; Medard, Y.; Azougagh, S.; Zhang, D.; Yakouben, K.; Jacqz-Aigrain, E. Determinants of mercaptopurine toxicity in paediatric acute lymphoblastic leukemia maintenance therapy. Br. J. Clin. Pharmacol. 2011, 71, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.B.; Szumlanski, C.; Weinshilboum, R.M.; Schmiegelow, K. Pharmacokinetics, dose adjustments, and 6-mercaptopurine/methotrexate drug interactions in two patients with thiopurine methyltransferase deficiency. Acta Paediatr. 1998, 87, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Ando, Y.; Hasegawa, Y.; Sekido, Y.; Shimokata, K.; Horibe, K. Genetic polymorphisms of thiopurine S-methyltransferase and 6-mercaptopurine toxicity in Japanese children with acute lymphoblastic leukaemia. Pharmacogenetics 2001, 11, 269–273. [Google Scholar] [CrossRef] [PubMed]
- De Ridder, L.; Van Dieren, J.M.; Van Deventer, H.J.; Stokkers, P.C.; Van der Woude, J.C.; Van Vuuren, A.J.; Benninga, M.A.; Escher, J.C.; Hommes, D.W. Pharmacogenetics of thiopurine therapy in paediatric IBD patients. Aliment. Pharmacol. Ther. 2006, 23, 1137–1141. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, L.; Urosevic, J.; Janic, D.; Jovanovic, N.; Petrucev, B.; Tosic, N.; Pavlovic, S. Analysis of thiopurine S-methyltransferase polymorphism in the population of Serbia and Montenegro and mercaptopurine therapy tolerance in childhood acute lymphoblastic leukemia. Ther. Drug Monit. 2006, 28, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Dubinsky, M.C.; Lamothe, S.; Yang, H.Y.; Targan, S.R.; Sinnett, D.; Theoret, Y.; Seidman, E.G. Pharmacogenomics and metabolite measurement for 6-mercaptopurine therapy in inflammatory bowel disease. Gastroenterology 2000, 118, 705–713. [Google Scholar] [CrossRef]
- Evans, W.E.; Horner, M.; Chu, Y.Q.; Kalwinsky, D.; Roberts, W.M. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J. Pediatr. 1991, 119, 985–989. [Google Scholar] [CrossRef]
- Ganiere-Monteil, C.; Medard, Y.; Lejus, C.; Bruneau, B.; Pineau, A.; Fenneteau, O.; Bourin, M.; Jacqz-Aigrain, E. Phenotype and genotype for thiopurine methyltransferase activity in the French Caucasian population: Impact of age. Eur. J. Clin. Pharmacol. 2004, 60, 89–96. [Google Scholar] [PubMed]
- Gazouli, M.; Pachoula, I.; Panayotou, I.; Mantzaris, G.; Syriopoulou, V.P.; Goutas, N.; Vlachodimitropoulos, D.; Anagnou, N.P.; Roma-Giannikou, E. Thiopurine S-methyltransferase genotype and the use of thiopurines in paediatric inflammatory bowel disease Greek patients. J. Clin. Pharm. Ther. 2010, 35, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Hindorf, U.; Peterson, C.; Almer, S. Assessment of thiopurine methyltransferase and metabolite formation during thiopurine therapy: Results from a large Swedish patient population. Ther. Drug Monit. 2004, 26, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, G.; Sinha, R.; Naithani, R.; Chandgothia, M. Thiopurine S-methyltransferase gene polymorphism and 6-mercaptopurine dose intensity in Indian children with acute lymphoblastic leukemia. Leuk. Res. 2010, 34, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Karas-Kuzelicki, N.; Jazbec, J.; Milek, M.; Mlinaric-Rascan, I. Heterozygosity at the TPMT gene locus, augmented by mutated MTHFR gene, predisposes to 6-MP related toxicities in childhood ALL patients. Leukemia 2009, 23, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Kham, S.K.; Tan, P.L.; Tay, A.H.; Heng, C.K.; Yeoh, A.E.; Quah, T.C. Thiopurine methyltransferase polymorphisms in a multiracial asian population and children with acute lymphoblastic leukemia. J. Pediatr. Hematol. Oncol. 2002, 24, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kang, H.J.; Kim, H.J.; Jang, M.K.; Kim, N.H.; Oh, Y.; Han, B.D.; Choi, J.Y.; Kim, C.W.; Lee, J.W.; et al. Pharmacogenetic analysis of pediatric patients with acute lymphoblastic leukemia: A possible association between survival rate and ITPA polymorphism. PLoS ONE 2012, 7, e45558. [Google Scholar] [CrossRef] [PubMed]
- Lennard, L.; Cartwright, C.S.; Wade, R.; Richards, S.M.; Vora, A. Thiopurine methyltransferase genotype-phenotype discordance and thiopurine active metabolite formation in childhood acute lymphoblastic leukaemia. Br. J. Clin. Pharmacol. 2013, 76, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Lennard, L.; Cartwright, C.S.; Wade, R.; Vora, A. Thiopurine dose intensity and treatment outcome in childhood lymphoblastic leukaemia: The influence of thiopurine methyltransferase pharmacogenetics. Br. J. Haematol. 2015, 169, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Lennard, L.; Gibson, B.E.; Nicole, T.; Lilleyman, J.S. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch. Dis. Child. 1993, 69, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Lennard, L.; Richards, S.; Cartwright, C.S.; Mitchell, C.; Lilleyman, J.S.; Vora, A. The thiopurine methyltransferase genetic polymorphism is associated with thioguanine-related veno-occlusive disease of the liver in children with acute lymphoblastic leukemia. Clin. Pharmacol. Ther. 2006, 80, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Levinsen, M.; Rosthoj, S.; Nygaard, U.; Heldrup, J.; Harila-Saari, A.; Jonsson, O.G.; Bechensteen, A.G.; Abrahamsson, J.; Lausen, B.; Frandsen, T.L.; et al. Myelotoxicity after high-dose methotrexate in childhood acute leukemia is influenced by 6-mercaptopurine dosing but not by intermediate thiopurine methyltransferase activity. Cancer Chemother. Pharmacol. 2015, 75, 59–66. [Google Scholar] [CrossRef] [PubMed]
- McBride, K.L.; Gilchrist, G.S.; Smithson, W.A.; Weinshilboum, R.M.; Szumlanski, C.L. Severe 6-thioguanine-induced marrow aplasia in a child with acute lymphoblastic leukemia and inherited thiopurine methyltransferase deficiency. J. Pediatr. Hematol. Oncol. 2000, 22, 441–445. [Google Scholar] [CrossRef] [PubMed]
- McLeod, H.L.; Coulthard, S.; Thomas, A.E.; Pritchard, S.C.; King, D.J.; Richards, S.M.; Eden, O.B.; Hall, A.G.; Gibson, B.E. Analysis of thiopurine methyltransferase variant alleles in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 1999, 105, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Peregud-Pogorzelski, J.; Tetera-Rudnicka, E.; Kurzawski, M.; Brodkiewicz, A.; Adrianowska, N.; Mlynarski, W.; Januszkiewicz, D.; Drozdzik, M. Thiopurine S-methyltransferase (TPMT) polymorphisms in children with acute lymphoblastic leukemia, and the need for reduction or cessation of 6-mercaptopurine doses during maintenance therapy: The Polish multicenter analysis. Pediatr. Blood Cancer 2011, 57, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Relling, M.V.; Hancock, M.L.; Rivera, G.K.; Sandlund, J.T.; Ribeiro, R.C.; Krynetski, E.Y.; Pui, C.H.; Evans, W.E. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J. Natl. Cancer Inst. 1999, 91, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Schaeffeler, E.; Stanulla, M.; Greil, J.; Schrappe, M.; Eichelbaum, M.; Zanger, U.M.; Schwab, M. A novel TPMT missense mutation associated with TPMT deficiency in a 5-year-old boy with ALL. Leukemia 2003, 17, 1422–1424. [Google Scholar] [CrossRef] [PubMed]
- Schmiegelow, K.; Forestier, E.; Kristinsson, J.; Soderhall, S.; Vettenranta, K.; Weinshilboum, R.; Wesenberg, F.; Nordic Society of Paediatric Haematology and Oncology. Thiopurine methyltransferase activity is related to the risk of relapse of childhood acute lymphoblastic leukemia: Results from the NOPHO ALL-92 study. Leukemia 2009, 23, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.R.; de Oliveira, B.M.; Viana, M.B.; Murao, M.; Romanha, A.J. Thiopurine S-methyltransferase (TPMT) gene polymorphism in Brazilian children with acute lymphoblastic leukemia: Association with clinical and laboratory data. Ther. Drug Monit. 2008, 30, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Stanulla, M.; Schaeffeler, E.; Flohr, T.; Cario, G.; Schrauder, A.; Zimmermann, M.; Welte, K.; Ludwig, W.D.; Bartram, C.R.; Zanger, U.M.; et al. Thiopurine methyltransferase (TPMT) genotype and early treatment response to mercaptopurine in childhood acute lymphoblastic leukemia. JAMA 2005, 293, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Stanulla, M.; Schaeffeler, E.; Moricke, A.; Coulthard, S.A.; Cario, G.; Schrauder, A.; Kaatsch, P.; Dordelmann, M.; Welte, K.; Zimmermann, M.; et al. Thiopurine methyltransferase genetics is not a major risk factor for secondary malignant neoplasms after treatment of childhood acute lymphoblastic leukemia on Berlin-Frankfurt-Munster protocols. Blood 2009, 114, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Stocco, G.; Martelossi, S.; Barabino, A.; Fontana, M.; Lionetti, P.; Decorti, G.; Malusa, N.; Bartoli, F.; Fezzi, M.; Giraldi, T.; et al. TPMT genotype and the use of thiopurines in paediatric inflammatory bowel disease. Dig. Liver Dis. 2005, 37, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Tumer, T.B.; Ulusoy, G.; Adali, O.; Sahin, G.; Gozdasoglu, S.; Arinc, E. The low frequency of defective TPMT alleles in Turkish population: A study on pediatric patients with acute lymphoblastic leukemia. Am. J. Hematol. 2007, 82, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Gao, X.; Ding, L.; Liu, H.; Wang, X.; Chen, B.; Bi, H.; Xiao, Y.; Cheng, P.; Zhao, L.; et al. Prospective Evaluation of Pharmacogenomics and Metabolite Measurements upon Azathioprine Therapy in Inflammatory Bowel Disease: An Observational Study. Medicine (Baltimore) 2016, 95, e3326. [Google Scholar]
- Chiengthong, K.; Ittiwut, C.; Muensri, S.; Sophonphan, J.; Sosothikul, D.; Seksan, P.; Suppipat, K.; Suphapeetiporn, K.; Shotelersuk, V. NUDT15 c.415C>T increases risk of 6-mercaptopurine induced myelosuppression during maintenance therapy in children with acute lymphoblastic leukemia. Haematologica 2016, 101, e24–e26. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Hwang, E.H.; Park, J.H.; Shin, J.H.; Kang, B.; Kim, S.Y. NUDT15 variant is the most common variant associated with thiopurine-induced early leukopenia and alopecia in Korean pediatric patients with Crohn’s disease. Eur. J. Gastroenterol. Hepatol. 2016, 28, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.C.; Yang, C.P.; Liu, H.C.; Jaing, T.H.; Chen, S.H.; Hung, I.J.; Yeh, T.C.; Lin, T.H.; Lai, C.L.; Lai, C.Y.; et al. NUDT15 gene polymorphism related to mercaptopurine intolerance in Taiwan Chinese children with acute lymphoblastic leukemia. Pharmacogenom. J. 2015. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, T.; Nishii, R.; Perez-Andreu, V.; Yang, W.; Klussmann, F.A.; Zhao, X.; Lin, T.N.; Hoshitsuki, K.; Nersting, J.; Kihira, K.; et al. NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat. Genet. 2016, 48, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Fukushima, H.; Suzuki, R.; Hosaka, S.; Yamaki, Y.; Kobayashi, C.; Sakai, A.; Imagawa, K.; Iwabuchi, A.; Yoshimi, A.; et al. Genotyping NUDT15 can predict the dose reduction of 6-MP for children with acute lymphoblastic leukemia especially at a preschool age. J. Hum. Genet. 2016. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kato, M.; Hasegawa, D.; Urayama, K.Y.; Nakadate, H.; Kondoh, K.; Nakamura, K.; Koh, K.; Komiyama, T.; Manabe, A. Susceptibility to 6-MP toxicity conferred by a NUDT15 variant in Japanese children with acute lymphoblastic leukaemia. Br. J. Haematol. 2015, 171, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Landier, W.; Yang, W.; Liu, C.; Hageman, L.; Cheng, C.; Pei, D.; Chen, Y.; Crews, K.R.; Kornegay, N.; et al. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J. Clin. Oncol. 2015, 33, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Ailing, Z.; Jing, Y.; Jingli, L.; Yun, X.; Xiaojian, Z. Further evidence that a variant of the gene NUDT15 may be an important predictor of azathioprine-induced toxicity in Chinese subjects: A case report. J. Clin. Pharm. Ther. 2016, 41, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Asada, A.; Nishida, A.; Shioya, M.; Imaeda, H.; Inatomi, O.; Bamba, S.; Kito, K.; Sugimoto, M.; Andoh, A. NUDT15 R139C-related thiopurine leukocytopenia is mediated by 6-thioguanine nucleotide-independent mechanism in Japanese patients with inflammatory bowel disease. J. Gastroenterol. 2016, 51, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Kakuta, Y.; Naito, T.; Onodera, M.; Kuroha, M.; Kimura, T.; Shiga, H.; Endo, K.; Negoro, K.; Kinouchi, Y.; Shimosegawa, T. NUDT15 R139C causes thiopurine-induced early severe hair loss and leukopenia in Japanese patients with IBD. Pharmacogenom. J. 2015. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Paradkar, M.; Desai, D.; Ashavaid, T.F. Nudt15 C415t Variant as a Predictor For Thiopurine Induced Toxicity in Indian Patients. J. Gastroenterol. Hepatol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.C.; Leung, A.W.; Kwok, J.S.; Chan, M.H.; Li, C.K.; Yuen, Y.P. NUDT15 variant and thiopurine-induced leukopenia in Hong Kong. Hong Kong Med. J. 2016, 22, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.K.; Hong, M.; Baek, J.; Choi, H.; Zhao, W.; Jung, Y.; Haritunians, T.; Ye, B.D.; Kim, K.J.; Park, S.H.; et al. A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nat. Genet. 2014, 46, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Caronia, D.; Patino-Garcia, A.; Milne, R.L.; Zalacain-Diez, M.; Pita, G.; Alonso, M.R.; Moreno, L.T.; Sierrasesumaga-Ariznabarreta, L.; Benitez, J.; Gonzalez-Neira, A. Common variations in ERCC2 are associated with response to cisplatin chemotherapy and clinical outcome in osteosarcoma patients. Pharmacogenom. J. 2009, 9, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Sakano, S.; Hinoda, Y.; Sasaki, M.; Wada, T.; Matsumoto, H.; Eguchi, S.; Shinohara, A.; Kawai, Y.; Hara, T.; Nagao, K.; et al. Nucleotide excision repair gene polymorphisms may predict acute toxicity in patients treated with chemoradiotherapy for bladder cancer. Pharmacogenomics 2010, 11, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.A.; Kulke, M.H.; Heist, R.S.; Zhou, W.; Ma, C.; Xu, W.; Marshall, A.L.; Zhai, R.; Hooshmand, S.M.; Asomaning, K.; et al. Cisplatin pharmacogenetics, DNA repair polymorphisms, and esophageal cancer outcomes. Pharmacogenet. Genom. 2009, 19, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Giovannetti, E.; Pacetti, P.; Reni, M.; Leon, L.G.; Mambrini, A.; Vasile, E.; Ghidini, M.; Funel, N.; Lucchesi, M.; Cereda, S.; et al. Association between DNA-repair polymorphisms and survival in pancreatic cancer patients treated with combination chemotherapy. Pharmacogenomics 2011, 12, 1641–1652. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.Y.; Huang, M.L.; Chen, M.J.; Lu, C.Y.; Chen, C.F.; Tsai, P.C.; Chuang, S.C.; Hou, M.F.; Lin, S.R.; Wang, J.Y. Multiple genetic polymorphisms in the prediction of clinical outcome of metastatic colorectal cancer patients treated with first-line FOLFOX-4 chemotherapy. Pharmacogenet. Genom. 2011, 21, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Kalikaki, A.; Kanaki, M.; Vassalou, H.; Souglakos, J.; Voutsina, A.; Georgoulias, V.; Mavroudis, D. DNA repair gene polymorphisms predict favorable clinical outcome in advanced non-small-cell lung cancer. Clin. Lung Cancer 2009, 10, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Khrunin, A.; Ivanova, F.; Moisseev, A.; Khokhrin, D.; Sleptsova, Y.; Gorbunova, V.; Limborska, S. Pharmacogenomics of cisplatin-based chemotherapy in ovarian cancer patients of different ethnic origins. Pharmacogenomics 2012, 13, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Khrunin, A.V.; Moisseev, A.; Gorbunova, V.; Limborska, S. Genetic polymorphisms and the efficacy and toxicity of cisplatin-based chemotherapy in ovarian cancer patients. Pharmacogenom. J. 2010, 10, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Stoehlmacher, J.; Park, D.J.; Zhang, W.; Yang, D.; Groshen, S.; Zahedy, S.; Lenz, H.J. A multivariate analysis of genomic polymorphisms: Prediction of clinical outcome to 5-FU/oxaliplatin combination chemotherapy in refractory colorectal cancer. Br. J. Cancer 2004, 91, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, I.; Salazar, J.; Majem, M.; Pallares, C.; Del Rio, E.; Paez, D.; Baiget, M.; Barnadas, A. Pharmacogenetics of the DNA repair pathways in advanced non-small cell lung cancer patients treated with platinum-based chemotherapy. Cancer Lett. 2014, 353, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Tzvetkov, M.V.; Behrens, G.; O'Brien, V.P.; Hohloch, K.; Brockmoller, J.; Benohr, P. Pharmacogenetic analyses of cisplatin-induced nephrotoxicity indicate a renoprotective effect of ERCC1 polymorphisms. Pharmacogenomics 2011, 12, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Shu-Ying, Y.; Shan, K.; Yip, B.H.; Rong-Miao, Z.; Na, W.; Hai-Yan, S. Association between polymorphisms of ERCC1 and survival in epithelial ovarian cancer patients with chemotherapy. Pharmacogenomics 2012, 13, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Zucali, P.A.; Giovannetti, E.; Destro, A.; Mencoboni, M.; Ceresoli, G.L.; Gianoncelli, L.; Lorenzi, E.; De Vincenzo, F.; Simonelli, M.; Perrino, M.; et al. Thymidylate synthase and excision repair cross-complementing group-1 as predictors of responsiveness in mesothelioma patients treated with pemetrexed/carboplatin. Clin. Cancer Res. 2011, 17, 2581–2590. [Google Scholar] [CrossRef] [PubMed]
- Perez-Ramirez, C.; Canadas-Garre, M.; Alnatsha, A.; Villar, E.; Delgado, J.R.; Faus-Dader, M.J.; Calleja-Hernandez, M.A. Pharmacogenetic predictors of toxicity to platinum based chemotherapy in non-small cell lung cancer patients. Pharmacol. Res. 2016, 111, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Tibaldi, C.; Giovannetti, E.; Vasile, E.; Mey, V.; Laan, A.C.; Nannizzi, S.; di Marsico, R.; Antonuzzo, A.; Orlandini, C.; Ricciardi, S.; et al. Correlation of CDA, ERCC1, and XPD polymorphisms with response and survival in gemcitabine/cisplatin-treated advanced non-small cell lung cancer patients. Clin. Cancer Res. 2008, 14, 1797–1803. [Google Scholar] [CrossRef] [PubMed]
- Abbas, M.; Kushwaha, V.S.; Srivastava, K.; Banerjee, M. Glutathione S-Transferase Gene Polymorphisms and Treatment Outcome in Cervical Cancer Patients under Concomitant Chemoradiation. PLoS ONE 2015, 10, e0142501. [Google Scholar] [CrossRef] [PubMed]
- Kap, E.J.; Richter, S.; Rudolph, A.; Jansen, L.; Ulrich, A.; Hoffmeister, M.; Ulrich, C.M.; Brenner, H.; Chang-Claude, J. Genetic variants in the glutathione S-transferase genes and survival in colorectal cancer patients after chemotherapy and differences according to treatment with oxaliplatin. Pharmacogenet. Genom. 2014, 24, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.; Assis, J.; Gomes, M.; Nogueira, A.; Medeiros, R. Improvement of a predictive model in ovarian cancer patients submitted to platinum-based chemotherapy: Implications of a GST activity profile. Eur. J. Clin. Pharmacol. 2016, 72, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Henriquez-Hernandez, L.A.; Murias-Rosales, A.; Gonzalez-Hernandez, A.; de Leon, A.C.; Diaz-Chico, N.; Fernandez-Perez, L. Distribution of TYMS, MTHFR, p53 and MDR1 gene polymorphisms in patients with breast cancer treated with neoadjuvant chemotherapy. Cancer Epidemiol. 2010, 34, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.H.; Hua, D.; Li, L.H.; Zhu, J.D. Prognostic role of p53 codon 72 polymorphism in gastric cancer patients treated with fluorouracil-based adjuvant chemotherapy. J. Cancer Res. Clin. Oncol. 2008, 134, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Sohn, S.K.; Chae, Y.S.; Song, H.S.; Kwon, K.Y.; Do, Y.R.; Kim, M.K.; Lee, K.H.; Hyun, M.S.; Lee, W.S.; et al. TP53 codon 72 polymorphism associated with prognosis in patients with advanced gastric cancer treated with paclitaxel and cisplatin. Cancer Chemother. Pharmacol. 2009, 64, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.H.; Kim, M.K.; Kim, J.W.; Park, N.H.; Song, Y.S.; Kang, S.B.; Lee, H.P. XRCC1 R399Q polymorphism is associated with response to platinum-based neoadjuvant chemotherapy in bulky cervical cancer. Gynecol. Oncol. 2006, 103, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Xie, X.; Wang, H.; Hu, J.; Liu, F.; Liu, Z.; Zhou, J.; Zhang, Y.; Xi, X.; Hu, B.; et al. ERCC1 Cys8092Ala and XRCC1 Arg399Gln polymorphisms predict progression-free survival after curative radiotherapy for nasopharyngeal carcinoma. PLoS ONE 2014, 9, e101256. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.Y.; Huang, Q.; Zhao, Y.C.; Zhou, H.H.; Liu, Z.Q. Meta-analysis on pharmacogenetics of platinum-based chemotherapy in non small cell lung cancer (NSCLC) patients. PLoS ONE 2012, 7, e38150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ma, W.; Li, Y.; Wu, J.; Shi, G.Y. Pharmacogenetics of DNA repair gene polymorphisms in non-small-cell lung carcinoma patients on platinum-based chemotherapy. Genet. Mol. Res. 2014, 13, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Ichinose, Y.; Ohe, Y.; Yamamoto, N.; Negoro, S.; Nishio, K.; Itoh, Y.; Jiang, H.; Duffield, E.; McCormack, R.; et al. Epidermal growth factor receptor mutation status in circulating free DNA in serum: From IPASS, a phase III study of gefitinib or carboplatin/paclitaxel in non-small cell lung cancer. J. Thorac. Oncol. 2012, 7, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Park, K.; Kim, S.W.; Lee, D.H.; Kim, H.Y.; Kim, H.T.; Ahn, M.J.; Yun, T.; Ahn, J.S.; Suh, C.; et al. First-SIGNAL: First-line single-agent iressa versus gemcitabine and cisplatin trial in never-smokers with adenocarcinoma of the lung. J. Clin. Oncol. 2012, 30, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Oizumi, S.; Kobayashi, K.; Inoue, A.; Maemondo, M.; Sugawara, S.; Yoshizawa, H.; Isobe, H.; Harada, M.; Kinoshita, I.; Okinaga, S.; et al. Quality of life with gefitinib in patients with EGFR-mutated non-small cell lung cancer: Quality of life analysis of North East Japan Study Group 002 Trial. Oncologist 2012, 17, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Cui, L.H.; Yu, Z.; Zhang, T.T.; Shin, M.H.; Kim, H.N.; Choi, J.S. Influence of polymorphisms in MTHFR 677 C→T, TYMS 3R→2R and MTR 2756 A→G on NSCLC risk and response to platinum-based chemotherapy in advanced NSCLC. Pharmacogenomics 2011, 12, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Smit, E.F.; Burgers, S.A.; Biesma, B.; Smit, H.J.; Eppinga, P.; Dingemans, A.M.; Joerger, M.; Schellens, J.H.; Vincent, A.; van Zandwijk, N.; et al. Randomized phase II and pharmacogenetic study of pemetrexed compared with pemetrexed plus carboplatin in pretreated patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2009, 27, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Gregers, J.; Green, H.; Christensen, I.J.; Dalhoff, K.; Schroeder, H.; Carlsen, N.; Rosthoej, S.; Lausen, B.; Schmiegelow, K.; Peterson, C. Polymorphisms in the ABCB1 gene and effect on outcome and toxicity in childhood acute lymphoblastic leukemia. Pharmacogenom. J. 2015, 15, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Suthandiram, S.; Gan, G.G.; Zain, S.M.; Bee, P.C.; Lian, L.H.; Chang, K.M.; Ong, T.C.; Mohamed, Z. Effect of polymorphisms within methotrexate pathway genes on methotrexate toxicity and plasma levels in adults with hematological malignancies. Pharmacogenomics 2014, 15, 1479–1494. [Google Scholar] [CrossRef] [PubMed]
- Zgheib, N.K.; Akra-Ismail, M.; Aridi, C.; Mahfouz, R.; Abboud, M.R.; Solh, H.; Muwakkit, S.A. Genetic polymorphisms in candidate genes predict increased toxicity with methotrexate therapy in Lebanese children with acute lymphoblastic leukemia. Pharmacogenet. Genom. 2014, 24, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Iannaccone, C.K.; Lee, Y.C.; Cui, J.; Frits, M.L.; Glass, R.J.; Plenge, R.M.; Solomon, D.H.; Weinblatt, M.E.; Shadick, N.A. Using genetic and clinical data to understand response to disease-modifying anti-rheumatic drug therapy: Data from the Brigham and Women’s Hospital Rheumatoid Arthritis Sequential Study. Rheumatology 2011, 50, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Cui, J.; Costenbader, K.H.; Shadick, N.A.; Weinblatt, M.E.; Karlson, E.W. Investigation of candidate polymorphisms and disease activity in rheumatoid arthritis patients on methotrexate. Rheumatology 2009, 48, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Aplenc, R.; Thompson, J.; Han, P.; La, M.; Zhao, H.; Lange, B.; Rebbeck, T. Methylenetetrahydrofolate reductase polymorphisms and therapy response in pediatric acute lymphoblastic leukemia. Cancer Res. 2005, 65, 2482–2487. [Google Scholar] [CrossRef] [PubMed]
- Araoz, H.V.; D'Aloi, K.; Foncuberta, M.E.; Sanchez La Rosa, C.G.; Alonso, C.N.; Chertkoff, L.; Felice, M. Pharmacogenetic studies in children with acute lymphoblastic leukemia in Argentina. Leuk. Lymphoma 2015, 56, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Costea, I.; Moghrabi, A.; Laverdiere, C.; Graziani, A.; Krajinovic, M. Folate cycle gene variants and chemotherapy toxicity in pediatric patients with acute lymphoblastic leukemia. Haematologica 2006, 91, 1113–1116. [Google Scholar] [PubMed]
- D'Angelo, V.; Ramaglia, M.; Iannotta, A.; Francese, M.; Pota, E.; Affinita, M.C.; Pecoraro, G.; Indolfi, C.; di Martino, M.; di Pinto, D.; et al. Influence of methylenetetrahydrofolate reductase gene polymorphisms on the outcome of pediatric patients with non-Hodgkin lymphoma treated with high-dose methotrexate. Leuk. Lymphoma 2013, 54, 2639–2644. [Google Scholar] [CrossRef] [PubMed]
- Dorababu, P.; Naushad, S.M.; Linga, V.G.; Gundeti, S.; Nagesh, N.; Kutala, V.K.; Reddanna, P.; Digumarti, R. Genetic variants of thiopurine and folate metabolic pathways determine 6-MP-mediated hematological toxicity in childhood ALL. Pharmacogenomics 2012, 13, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- El-Khodary, N.M.; El-Haggar, S.M.; Eid, M.A.; Ebeid, E.N. Study of the pharmacokinetic and pharmacogenetic contribution to the toxicity of high-dose methotrexate in children with acute lymphoblastic leukemia. Med. Oncol. 2012, 29, 2053–2062. [Google Scholar] [CrossRef] [PubMed]
- Hagleitner, M.M.; Coenen, M.J.; Aplenc, R.; Patino-Garcia, A.; Chiusolo, P.; Gemmati, D.; de Mattei, M.; Ongaro, A.; Krajinovic, M.; Hoogerbrugge, P.M.; et al. The role of the MTHFR 677C>T polymorphism in methotrexate-induced liver toxicity: A meta-analysis in patients with cancer. Pharmacogenom. J. 2014, 14, 115–119. [Google Scholar] [CrossRef] [PubMed]
- He, H.R.; Chen, S.Y.; You, H.S.; Hu, S.S.; Sun, J.Y.; Dong, Y.L.; Lu, J. Association between methylenetetrahydrofolate reductase polymorphisms and the relapse of acute lymphoblastic leukemia: A meta-analysis. Pharmacogenom. J. 2014, 14, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, H.; Okamura, N.; Yagi, M.; Noro, Y.; Moriya, Y.; Nakamura, T.; Hayakawa, A.; Takeshima, Y.; Sakaeda, T.; Matsuo, M.; et al. Genetic polymorphisms associated with adverse events and elimination of methotrexate in childhood acute lymphoblastic leukemia and malignant lymphoma. J. Hum. Genet. 2007, 52, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Jazbec, J.; Kitanovski, L.; Aplenc, R.; Debeljak, M.; Dolzan, V. No evidence of association of methylenetetrahydrofolate reductase polymorphism with occurrence of second neoplasms after treatment of childhood leukemia. Leuk. Lymphoma 2005, 46, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Kishi, S.; Griener, J.; Cheng, C.; Das, S.; Cook, E.H.; Pei, D.; Hudson, M.; Rubnitz, J.; Sandlund, J.T.; Pui, C.H.; et al. Homocysteine, pharmacogenetics, and neurotoxicity in children with leukemia. J. Clin. Oncol. 2003, 21, 3084–3091. [Google Scholar] [CrossRef] [PubMed]
- Krajinovic, M.; Lemieux-Blanchard, E.; Chiasson, S.; Primeau, M.; Costea, I.; Moghrabi, A. Role of polymorphisms in MTHFR and MTHFD1 genes in the outcome of childhood acute lymphoblastic leukemia. Pharmacogenom. J. 2004, 4, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Krajinovic, M.; Robaey, P.; Chiasson, S.; Lemieux-Blanchard, E.; Rouillard, M.; Primeau, M.; Bournissen, F.G.; Moghrabi, A. Polymorphisms of genes controlling homocysteine levels and IQ score following the treatment for childhood ALL. Pharmacogenomics 2005, 6, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, E.; Martin-Guerrero, I.; Ballesteros, J.; Garcia-Orad, A. A systematic review and meta-analysis of MTHFR polymorphisms in methotrexate toxicity prediction in pediatric acute lymphoblastic leukemia. Pharmacogenom. J. 2013, 13, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Pakakasama, S.; Kanchanakamhaeng, K.; Kajanachumpol, S.; Udomsubpayakul, U.; Sirachainan, N.; Thithapandha, A.; Hongeng, S. Genetic polymorphisms of folate metabolic enzymes and toxicities of high dose methotrexate in children with acute lymphoblastic leukemia. Ann. Hematol. 2007, 86, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Patino-Garcia, A.; Zalacain, M.; Marrodan, L.; San-Julian, M.; Sierrasesumaga, L. Methotrexate in pediatric osteosarcoma: Response and toxicity in relation to genetic polymorphisms and dihydrofolate reductase and reduced folate carrier 1 expression. J. Pediatri. 2009, 154, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Roy Moulik, N.; Kumar, A.; Agrawal, S.; Awasthi, S.; Mahdi, A.A.; Kumar, A. Role of folate status and methylenetetrahydrofolate reductase genotype on the toxicity and outcome of induction chemotherapy in children with acute lymphoblastic leukemia. Leuk. Lymphoma 2015, 56, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.; Altes, A.; del Rio, E.; Estella, J.; Rives, S.; Tasso, M.; Navajas, A.; Molina, J.; Villa, M.; Vivanco, J.L.; et al. Methotrexate consolidation treatment according to pharmacogenetics of MTHFR ameliorates event-free survival in childhood acute lymphoblastic leukaemia. Pharmacogenom. J. 2012, 12, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Seidemann, K.; Book, M.; Zimmermann, M.; Meyer, U.; Welte, K.; Stanulla, M.; Reiter, A. MTHFR 677 (C→T) polymorphism is not relevant for prognosis or therapy-associated toxicity in pediatric NHL: Results from 484 patients of multicenter trial NHL-BFM 95. Ann. Hematol. 2006, 85, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Shimasaki, N.; Mori, T.; Samejima, H.; Sato, R.; Shimada, H.; Yahagi, N.; Torii, C.; Yoshihara, H.; Tanigawara, Y.; Takahashi, T.; et al. Effects of methylenetetrahydrofolate reductase and reduced folate carrier 1 polymorphisms on high-dose methotrexate-induced toxicities in children with acute lymphoblastic leukemia or lymphoma. J. Pediatr. Hematol. Oncol. 2006, 28, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Shimasaki, N.; Mori, T.; Torii, C.; Sato, R.; Shimada, H.; Tanigawara, Y.; Kosaki, K.; Takahashi, T. Influence of MTHFR and RFC1 polymorphisms on toxicities during maintenance chemotherapy for childhood acute lymphoblastic leukemia or lymphoma. J. Pediatr. Hematol. Oncol. 2008, 30, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Spyridopoulou, K.P.; Dimou, N.L.; Hamodrakas, S.J.; Bagos, P.G. Methylene tetrahydrofolate reductase gene polymorphisms and their association with methotrexate toxicity: A meta-analysis. Pharmacogenet. Genom. 2012, 22, 117–133. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, S.I.; Yanagimachi, M.; Tanoshima, R.; Urayama, K.Y.; Tanaka, F.; Aida, N.; Goto, H.; Ito, S. Influence of ADORA2A gene polymorphism on leukoencephalopathy risk in MTX-treated pediatric patients affected by hematological malignancies. Pediatr. Blood Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- Yanagimachi, M.; Goto, H.; Kaneko, T.; Naruto, T.; Sasaki, K.; Takeuchi, M.; Tanoshima, R.; Kato, H.; Yokosuka, T.; Kajiwara, R.; et al. Influence of pre-hydration and pharmacogenetics on plasma methotrexate concentration and renal dysfunction following high-dose methotrexate therapy. Int. J. Hematol. 2013, 98, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Brooks, J.D.; Teraoka, S.N.; Bernstein, L.; Mellemkjaer, L.; Malone, K.E.; Lynch, C.F.; Haile, R.W.; Concannon, P.; Reiner, A.S.; Duggan, D.J.; et al. Common variants in genes coding for chemotherapy metabolizing enzymes, transporters, and targets: A case-control study of contralateral breast cancer risk in the WECARE Study. Cancer Causes Control. 2013, 24, 1605–1614. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, P.; Tulsyan, S.; Agarwal, G.; Lal, P.; Agrawal, S.; Mittal, R.D.; Mittal, B. Relationship of MTHFR and NQO1 Pharmacogenetics and Chemotherapy Clinical Outcomes in Breast Cancer Patients. Biochem. Genet. 2015, 53, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Chiusolo, P.; Reddiconto, G.; Casorelli, I.; Laurenti, L.; Sora, F.; Mele, L.; Annino, L.; Leone, G.; Sica, S. Preponderance of methylenetetrahydrofolate reductase C677T homozygosity among leukemia patients intolerant to methotrexate. Ann. Oncol. 2002, 13, 1915–1918. [Google Scholar] [CrossRef] [PubMed]
- Chiusolo, P.; Reddiconto, G.; Farina, G.; Mannocci, A.; Fiorini, A.; Palladino, M.; La Torre, G.; Fianchi, L.; Sora, F.; Laurenti, L.; et al. MTHFR polymorphisms' influence on outcome and toxicity in acute lymphoblastic leukemia patients. Leuk. Res. 2007, 31, 1669–1674. [Google Scholar] [CrossRef] [PubMed]
- Gemmati, D.; Ongaro, A.; Tognazzo, S.; Catozzi, L.; Federici, F.; Mauro, E.; della Porta, M.; Campioni, D.; Bardi, A.; Gilli, G.; et al. Methylenetetrahydrofolate reductase C677T and A1298C gene variants in adult non-Hodgkin’s lymphoma patients: Association with toxicity and survival. Haematologica 2007, 92, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Jabeen, S.; Holmboe, L.; Alnaes, G.I.; Andersen, A.M.; Hall, K.S.; Kristensen, V.N. Impact of genetic variants of RFC1, DHFR and MTHFR in osteosarcoma patients treated with high-dose methotrexate. Pharmacogenom. J. 2015, 15, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Ongaro, A.; De Mattei, M.; Della Porta, M.G.; Rigolin, G.; Ambrosio, C.; Di Raimondo, F.; Pellati, A.; Masieri, F.F.; Caruso, A.; Catozzi, L.; et al. Gene polymorphisms in folate metabolizing enzymes in adult acute lymphoblastic leukemia: Effects on methotrexate-related toxicity and survival. Haematologica 2009, 94, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Robien, K.; Schubert, M.M.; Bruemmer, B.; Lloid, M.E.; Potter, J.D.; Ulrich, C.M. Predictors of oral mucositis in patients receiving hematopoietic cell transplants for chronic myelogenous leukemia. J. Clin. Oncol. 2004, 22, 1268–1275. [Google Scholar] [CrossRef] [PubMed]
- Robien, K.; Schubert, M.M.; Chay, T.; Bigler, J.; Storb, R.; Yasui, Y.; Potter, J.D.; Ulrich, C.M. Methylenetetrahydrofolate reductase and thymidylate synthase genotypes modify oral mucositis severity following hematopoietic stem cell transplantation. Bone Marrow Transplant. 2006, 37, 799–800. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, C.M.; Yasui, Y.; Storb, R.; Schubert, M.M.; Wagner, J.L.; Bigler, J.; Ariail, K.S.; Keener, C.L.; Li, S.; Liu, H.; et al. Pharmacogenetics of methotrexate: Toxicity among marrow transplantation patients varies with the methylenetetrahydrofolate reductase C677T polymorphism. Blood 2001, 98, 231–234. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, R.; Hooijberg, J.H.; van Zelst, B.D.; Jansen, G.; van Zantwijk, C.H.; Kaspers, G.J.; Peters, G.J.; Ravindranath, Y.; Pieters, R.; Lindemans, J. Effect of polymorphisms in folate-related genes on in vitro methotrexate sensitivity in pediatric acute lymphoblastic leukemia. Blood 2005, 106, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Tissing, W.J.; de Jonge, R.; van Zelst, B.D.; Pieters, R. Polymorphisms in folate-related genes: Association with side effects of high-dose methotrexate in childhood acute lymphoblastic leukemia. Leukemia 2008, 22, 1798–1800. [Google Scholar] [CrossRef] [PubMed]
- Goricar, K.; Kovac, V.; Jazbec, J.; Zakotnik, B.; Lamovec, J.; Dolzan, V. Influence of the folate pathway and transporter polymorphisms on methotrexate treatment outcome in osteosarcoma. Pharmacogenet. Genom. 2014, 24, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, L.B.; Panetta, J.C.; Smith, C.; Yang, W.; Fan, Y.; Winick, N.J.; Martin, P.L.; Cheng, C.; Devidas, M.; Pui, C.H.; et al. Genome-wide study of methotrexate clearance replicates SLCO1B1. Blood 2013, 121, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Stocco, G.; Yang, W.; Crews, K.R.; Thierfelder, W.E.; Decorti, G.; Londero, M.; Franca, R.; Rabusin, M.; Valsecchi, M.G.; Pei, D.; et al. PACSIN2 polymorphism influences TPMT activity and mercaptopurine-related gastrointestinal toxicity. Hum. Mol. Genet. 2012, 21, 4793–4804. [Google Scholar] [CrossRef] [PubMed]
- Trevino, L.R.; Shimasaki, N.; Yang, W.; Panetta, J.C.; Cheng, C.; Pei, D.; Chan, D.; Sparreboom, A.; Giacomini, K.M.; Pui, C.H.; et al. Germline genetic variation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. J. Clin. Oncol. 2009, 27, 5972–5978. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.L.; Rodrigues, F.F.; Santos, R.E.; Aoki, T.; Rocha, M.N.; Longui, C.A.; Melo, M.B. GSTT1, GSTM1, and GSTP1 polymorphisms and chemotherapy response in locally advanced breast cancer. Genet. Mol. Res. 2010, 9, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.L.; Sun, T.; Zhang, B.N.; Zheng, S.; Lu, N.; Xu, B.H.; Wang, X.; Chen, G.J.; Yu, D.K.; Lin, D.X. Polymorphisms of GSTP1 is associated with differences of chemotherapy response and toxicity in breast cancer. Chin. Med. J. 2011, 124, 199–204. [Google Scholar] [PubMed]
- Glynn, S.A.; Boersma, B.J.; Howe, T.M.; Edvardsen, H.; Geisler, S.B.; Goodman, J.E.; Ridnour, L.A.; Lonning, P.E.; Borresen-Dale, A.L.; Naume, B.; et al. A mitochondrial target sequence polymorphism in manganese superoxide dismutase predicts inferior survival in breast cancer patients treated with cyclophosphamide. Clin. Cancer Res. 2009, 15, 4165–4173. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Shin, E.S.; Lee, Y.S.; Ghang, H.Y.; Kim, S.Y.; Hwang, J.A.; Kim, J.Y.; Lee, J.S. A genome-wide association study for irinotecan-related severe toxicities in patients with advanced non-small-cell lung cancer. Pharmacogenom. J. 2013, 13, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Saka, H.; Ando, M.; Sawa, T.; Muro, K.; Ueoka, H.; Yokoyama, A.; Saitoh, S.; Shimokata, K.; Hasegawa, Y. Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: A pharmacogenetic analysis. Cancer Res. 2000, 60, 6921–6926. [Google Scholar] [PubMed]
- Chen, Y.J.; Hu, F.; Li, C.Y.; Fang, J.M.; Chu, L.; Zhang, X.; Xu, Q. The association of UGT1A1*6 and UGT1A1*28 with irinotecan-induced neutropenia in Asians: A Meta-analysis. Biomarkers 2014, 19, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Li, M.; Hu, J.; Ren, W.; Xie, L.; Sun, Z.P.; Liu, B.R.; Xu, G.X.; Dong, X.L.; Qian, X.P. UGT1A1*6 polymorphisms are correlated with irinotecan-induced toxicity: A system review and meta-analysis in Asians. Cancer Chemother. Pharmacol. 2014, 73, 551–660. [Google Scholar] [CrossRef] [PubMed]
- Cote, J.F.; Kirzin, S.; Kramar, A.; Mosnier, J.F.; Diebold, M.D.; Soubeyran, I.; Thirouard, A.S.; Selves, J.; Laurent-Puig, P.; Ychou, M. UGT1A1 polymorphism can predict hematologic toxicity in patients treated with irinotecan. Clin. Cancer Res. 2007, 13, 3269–3275. [Google Scholar] [CrossRef] [PubMed]
- De Jong, F.A.; Kehrer, D.F.; Mathijssen, R.H.; Creemers, G.J.; de Bruijn, P.; van Schaik, R.H.; Planting, A.S.; van der Gaast, A.; Eskens, F.A.; Janssen, J.T.; et al. Prophylaxis of irinotecan-induced diarrhea with neomycin and potential role for UGT1A1*28 genotype screening: A double-blind, randomized, placebo-controlled study. Oncologist 2006, 11, 944–954. [Google Scholar] [CrossRef] [PubMed]
- De Jong, F.A.; Scott-Horton, T.J.; Kroetz, D.L.; McLeod, H.L.; Friberg, L.E.; Mathijssen, R.H.; Verweij, J.; Marsh, S.; Sparreboom, A. Irinotecan-induced diarrhea: Functional significance of the polymorphic ABCC2 transporter protein. Clin. Pharmacol. Ther. 2007, 81, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.M.; McKinnon, R.A.; Sorich, M.J. Impact of the UGT1A1*28 allele on response to irinotecan: A systematic review and meta-analysis. Pharmacogenomics 2012, 13, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.M.; Pignon, J.P.; Karapetis, C.S.; Boige, V.; Glimelius, B.; Kweekel, D.M.; Lara, P.N.; Laurent-Puig, P.; Martinez-Balibrea, E.; Paez, D.; et al. The effect of the UGT1A1*28 allele on survival after irinotecan-based chemotherapy: A collaborative meta-analysis. Pharmacogenom. J. 2014, 14, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Falvella, F.S.; Cheli, S.; Martinetti, A.; Mazzali, C.; Iacovelli, R.; Maggi, C.; Gariboldi, M.; Pierotti, M.A.; di Bartolomeo, M.; Sottotetti, E.; et al. DPD and UGT1A1 deficiency in colorectal cancer patients receiving triplet chemotherapy with fluoropyrimidines, oxaliplatin and irinotecan. Br. J. Clin. Pharmacol. 2015, 80, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Ferraldeschi, R.; Minchell, L.J.; Roberts, S.A.; Tobi, S.; Hadfield, K.D.; Blackhall, F.H.; Mullamitha, S.; Wilson, G.; Valle, J.; Saunders, M.; et al. UGT1A1*28 genotype predicts gastrointestinal toxicity in patients treated with intermediate-dose irinotecan. Pharmacogenomics 2009, 10, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Han, F.F.; Guo, C.L.; Yu, D.; Zhu, J.; Gong, L.L.; Li, G.R.; Lv, Y.L.; Liu, H.; An, G.Y.; Liu, L.H. Associations between UGT1A1*6 or UGT1A1*6/*28 polymorphisms and irinotecan-induced neutropenia in Asian cancer patients. Cancer Chemother. Pharmacol. 2014, 73, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Lim, H.S.; Park, Y.H.; Lee, S.Y.; Lee, J.S. Integrated pharmacogenetic prediction of irinotecan pharmacokinetics and toxicity in patients with advanced non-small cell lung cancer. Lung Cancer 2009, 63, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Lim, H.S.; Shin, E.S.; Yoo, Y.K.; Park, Y.H.; Lee, J.E.; Jang, I.J.; Lee, D.H.; Lee, J.S. Comprehensive analysis of UGT1A polymorphisms predictive for pharmacokinetics and treatment outcome in patients with non-small-cell lung cancer treated with irinotecan and cisplatin. J. Clin. Oncol. 2006, 24, 2237–2244. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, J.M.; Goldberg, R.M.; Qu, P.; Ibrahim, J.G.; McLeod, H.L. UGT1A1*28 genotype and irinotecan-induced neutropenia: Dose matters. J. Natl. Cancer Inst. 2007, 99, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.Y.; Yu, Q.; Pei, Q.; Guo, C. Dose-dependent association between UGT1A1*28 genotype and irinotecan-induced neutropenia: Low doses also increase risk. Clin. Cancer Res. 2010, 16, 3832–3842. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.Y.; Yu, Q.; Zhao, Y.S. Dose-dependent association between UGT1A1*28 polymorphism and irinotecan-induced diarrhoea: A meta-analysis. Eur. J. Cancer 2010, 46, 1856–1865. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, F.; Undevia, S.D.; Iyer, L.; Chen, P.X.; Das, S.; Kocherginsky, M.; Karrison, T.; Janisch, L.; Ramirez, J.; Rudin, C.M.; et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J. Clin. Oncol. 2004, 22, 1382–1388. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.; Das, S.; Janisch, L.; Wen, M.; Ramirez, J.; Karrison, T.; Fleming, G.F.; Vokes, E.E.; Schilsky, R.L.; Ratain, M.J. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenom. J. 2002, 2, 43–47. [Google Scholar] [CrossRef]
- Kim, S.Y.; Y, S.H.; E, K.S.; Kong, S.Y.; Shin, A.; Baek, J.Y.; Jung, K.H. S-1 plus irinotecan and oxaliplatin for the first-line treatment of patients with metastatic colorectal cancer: A prospective phase II study and pharmacogenetic analysis. Br. J. Cancer 2013, 109, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Kweekel, D.M.; Gelderblom, H.; Van der Straaten, T.; Antonini, N.F.; Punt, C.J.; Guchelaar, H.J. UGT1A1*28 genotype and irinotecan dosage in patients with metastatic colorectal cancer: A Dutch Colorectal Cancer Group study. Br. J. Cancer 2008, 99, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Lankisch, T.O.; Schulz, C.; Zwingers, T.; Erichsen, T.J.; Manns, M.P.; Heinemann, V.; Strassburg, C.P. Gilbert’s Syndrome and irinotecan toxicity: Combination with UDP-glucuronosyltransferase 1A7 variants increases risk. Cancer Epidemiol. Biomark. Prev. 2008, 17, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Levesque, E.; Belanger, A.S.; Harvey, M.; Couture, F.; Jonker, D.; Innocenti, F.; Cecchin, E.; Toffoli, G.; Guillemette, C. Refining the UGT1A haplotype associated with irinotecan-induced hematological toxicity in metastatic colorectal cancer patients treated with 5-fluorouracil/irinotecan-based regimens. J. Pharmacol. Exp. Ther. 2013, 345, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Z.; Guo, J.; Liu, J.; Li, C.; Liu, L.; Shi, H.; Liu, L.; Li, H.; Xie, C.; et al. Clinical significance of UGT1A1 gene polymorphisms on irinotecan-based regimens as the treatment in metastatic colorectal cancer. Onco Targets Ther. 2014, 7, 1653–1661. [Google Scholar] [PubMed]
- Liu, C.Y.; Chen, P.M.; Chiou, T.J.; Liu, J.H.; Lin, J.K.; Lin, T.C.; Chen, W.S.; Jiang, J.K.; Wang, H.S.; Wang, W.S. UGT1A1*28 polymorphism predicts irinotecan-induced severe toxicities without affecting treatment outcome and survival in patients with metastatic colorectal carcinoma. Cancer 2008, 112, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cheng, D.; Kuang, Q.; Liu, G.; Xu, W. Association of UGT1A1*28 polymorphisms with irinotecan-induced toxicities in colorectal cancer: A meta-analysis in Caucasians. Pharmacogenom. J. 2014, 14, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.Y.; Huang, X.E.; Wu, X.Y.; Cao, J.; Liu, J.; Wang, L.; Xiang, J. Clinical observations on associations between the UGT1A1 genotype and severe toxicity of irinotecan. Asian Pac. J. Cancer Prev. 2014, 15, 3335–3341. [Google Scholar] [CrossRef] [PubMed]
- Marcuello, E.; Altes, A.; Menoyo, A.; Del Rio, E.; Gomez-Pardo, M.; Baiget, M. UGT1A1 gene variations and irinotecan treatment in patients with metastatic colorectal cancer. Br. J. Cancer 2004, 91, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Massacesi, C.; Terrazzino, S.; Marcucci, F.; Rocchi, M.B.; Lippe, P.; Bisonni, R.; Lombardo, M.; Pilone, A.; Mattioli, R.; Leon, A. Uridine diphosphate glucuronosyl transferase 1A1 promoter polymorphism predicts the risk of gastrointestinal toxicity and fatigue induced by irinotecan-based chemotherapy. Cancer 2006, 106, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Onoue, M.; Terada, T.; Kobayashi, M.; Katsura, T.; Matsumoto, S.; Yanagihara, K.; Nishimura, T.; Kanai, M.; Teramukai, S.; Shimizu, A.; et al. UGT1A1*6 polymorphism is most predictive of severe neutropenia induced by irinotecan in Japanese cancer patients. Int. J. Clin. Oncol. 2009, 14, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Rouits, E.; Boisdron-Celle, M.; Dumont, A.; Guerin, O.; Morel, A.; Gamelin, E. Relevance of different UGT1A1 polymorphisms in irinotecan-induced toxicity: A molecular and clinical study of 75 patients. Clin. Cancer Res. 2004, 10, 5151–5159. [Google Scholar] [CrossRef] [PubMed]
- Rouits, E.; Charasson, V.; Petain, A.; Boisdron-Celle, M.; Delord, J.P.; Fonck, M.; Laurand, A.; Poirier, A.L.; Morel, A.; Chatelut, E.; et al. Pharmacokinetic and pharmacogenetic determinants of the activity and toxicity of irinotecan in metastatic colorectal cancer patients. Br. J. Cancer 2008, 99, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Ruzzo, A.; Graziano, F.; Loupakis, F.; Santini, D.; Catalano, V.; Bisonni, R.; Ficarelli, R.; Fontana, A.; Andreoni, F.; Falcone, A.; et al. Pharmacogenetic profiling in patients with advanced colorectal cancer treated with first-line FOLFIRI chemotherapy. Pharmacogenom. J. 2008, 8, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.F.; Panetta, J.C.; O’Shaughnessy, M.A.; Throm, S.L.; Fraga, C.H.; Owens, T.; Liu, T.; Billups, C.; Rodriguez-Galindo, C.; Gajjar, A.; et al. UGT1A1 promoter genotype correlates with SN-38 pharmacokinetics, but not severe toxicity in patients receiving low-dose irinotecan. J. Clin. Oncol. 2007, 25, 2594–2600. [Google Scholar] [CrossRef] [PubMed]
- Takano, M.; Kato, M.; Yoshikawa, T.; Sasaki, N.; Hirata, J.; Furuya, K.; Takahashi, M.; Yokota, H.; Kino, N.; Horie, K.; et al. Clinical significance of UDP-glucuronosyltransferase 1A1*6 for toxicities of combination chemotherapy with irinotecan and cisplatin in gynecologic cancers: A prospective multi-institutional study. Oncology 2009, 76, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Liu, Y.; Xi, W.Q.; Zhou, C.F.; Jiang, J.L.; Ma, T.; Ye, Z.B.; Zhang, J.; Zhu, Z.G. Relationship between UGT1A1*6/*28 polymorphisms and severe toxicities in Chinese patients with pancreatic or biliary tract cancer treated with irinotecan-containing regimens. Drug Des. Dev. Ther. 2015, 9, 3677–3683. [Google Scholar]
- Atasilp, C.; Chansriwong, P.; Sirachainan, E.; Reungwetwattana, T.; Chamnanphon, M.; Puangpetch, A.; Wongwaisayawan, S.; Sukasem, C. Correlation of UGT1A1*28 and *6 polymorphisms with irinotecan-induced neutropenia in Thai colorectal cancer patients. Drug Metab. Pharmacokinet. 2016, 31, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Takano, M.; Yamamoto, K.; Tabata, T.; Minegishi, Y.; Yokoyama, T.; Hirata, E.; Ikeda, T.; Shimada, M.; Yamada, K.; Morita, S.; et al. Impact of UGT1A1 genotype upon toxicities of combination with low-dose irinotecan plus platinum. Asia Pac. J. Clin. Oncol. 2016, 12, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Ding, Y.Y.; Song, L.X.; Xu, J.F. Correlation of UGT1A1 and ERCC1 gene polymorphisms with the outcome of combined irinotecan plus cisplatin treatment in recurrent ovarian cancer. Genet. Mol. Res. 2015, 14, 7241–7247. [Google Scholar] [CrossRef] [PubMed]
- Relling, M.V.; Gardner, E.E.; Sandborn, W.J.; Schmiegelow, K.; Pui, C.H.; Yee, S.W.; Stein, C.M.; Carrillo, M.; Evans, W.E.; Hicks, J.K.; et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin. Pharmacol. Ther. 2013, 89, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Relling, M.V.; Gardner, E.E.; Sandborn, W.J.; Schmiegelow, K.; Pui, C.H.; Yee, S.W.; Stein, C.M.; Carrillo, M.; Evans, W.E.; Klein, T.E. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin. Pharmacol. Ther. 2011, 93, 324–325. [Google Scholar] [CrossRef] [PubMed]
- Szumlanski, C.; Otterness, D.; Her, C.; Lee, D.; Brandriff, B.; Kelsell, D.; Spurr, N.; Lennard, L.; Wieben, E.; Weinshilboum, R. Thiopurine methyltransferase pharmacogenetics: Human gene cloning and characterization of a common polymorphism. DNA Cell Biol. 1996, 15, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Weinshilboum, R.M.; Sladek, S.L. Mercaptopurine pharmacogenetics: Monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am. J. Hum. Genet. 1980, 32, 651–662. [Google Scholar] [PubMed]
- Collie-Duguid, E.S.; Pritchard, S.C.; Powrie, R.H.; Sludden, J.; Collier, D.A.; Li, T.; McLeod, H.L. The frequency and distribution of thiopurine methyltransferase alleles in Caucasian and Asian populations. Pharmacogenetics 1999, 9, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Appell, M.L.; Berg, J.; Duley, J.; Evans, W.E.; Kennedy, M.A.; Lennard, L.; Marinaki, T.; McLeod, H.L.; Relling, M.V.; Schaeffeler, E.; et al. Nomenclature for alleles of the thiopurine methyltransferase gene. Pharmacogenet. Genom. 2013, 23, 242–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swen, J.J.; Nijenhuis, M.; de Boer, A.; Grandia, L.; Maitland-van der Zee, A.H.; Mulder, H.; Rongen, G.A.; van Schaik, R.H.; Schalekamp, T.; Touw, D.J.; et al. Pharmacogenetics: From bench to byte—An update of guidelines. Clin. Pharmacol. Ther. 2011, 89, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Zaza, G.; Cheok, M.; Krynetskaia, N.; Thorn, C.; Stocco, G.; Hebert, J.M.; McLeod, H.; Weinshilboum, R.M.; Relling, M.V.; Evans, W.E.; et al. Thiopurine pathway. Pharmacogenet. Genom. 2010, 20, 573. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.; Jemth, A.S.; Hagenkort, A.; Page, B.D.; Gustafsson, R.; Griese, J.J.; Gad, H.; Valerie, N.C.; Desroses, M.; Bostrom, J.; et al. Crystal structure, biochemical and cellular activities demonstrate separate functions of MTH1 and MTH2. Nat. Commun. 2015, 6, 7871. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, A.; Trombatore, G.; Triarico, S.; Arena, R.; Ferrara, P.; Scalzone, M.; Pierri, F.; Riccardi, R. Platinum compounds in children with cancer: Toxicity and clinical management. Anticancer Drugs 2013, 24, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.T.; Lipp, H.P. Toxicity of platinum compounds. Expert Opin. Pharmacother. 2003, 4, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Skinner, R.; Parry, A.; Price, L.; Cole, M.; Craft, A.W.; Pearson, A.D. Persistent nephrotoxicity during 10-year follow-up after cisplatin or carboplatin treatment in childhood: Relevance of age and dose as risk factors. Eur. J. Cancer 2009, 45, 3213–3219. [Google Scholar] [CrossRef] [PubMed]
- Zsiros, J.; Brugieres, L.; Brock, P.; Roebuck, D.; Maibach, R.; Zimmermann, A.; Childs, M.; Pariente, D.; Laithier, V.; Otte, J.B.; et al. Dose-dense cisplatin-based chemotherapy and surgery for children with high-risk hepatoblastoma (SIOPEL-4): A prospective, single-arm, feasibility study. Lancet. Oncol. 2013, 14, 834–842. [Google Scholar] [CrossRef]
- Karasawa, T.; Steyger, P.S. An integrated view of cisplatin-induced nephrotoxicity and ototoxicity. Toxicol. Lett. 2015, 237, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.V.; Heller, G. Chemotherapy dose intensity correlates strongly with response, median survival, and median progression-free survival in metastatic neuroblastoma. J. Clin. Oncol. 1991, 9, 1050–1058. [Google Scholar] [PubMed]
- Ross, C.J.; Katzov-Eckert, H.; Dube, M.P.; Brooks, B.; Rassekh, S.R.; Barhdadi, A.; Feroz-Zada, Y.; Visscher, H.; Brown, A.M.; Rieder, M.J.; et al. Genetic variants in TPMT and COMT are associated with hearing loss in children receiving cisplatin chemotherapy. Nat. Genet. 2009, 41, 1345–1349. [Google Scholar] [CrossRef] [PubMed]
- Hagleitner, M.M.; Coenen, M.J.; Patino-Garcia, A.; de Bont, E.S.; Gonzalez-Neira, A.; Vos, H.I.; van Leeuwen, F.N.; Gelderblom, H.; Hoogerbrugge, P.M.; Guchelaar, H.J.; et al. Influence of genetic variants in TPMT and COMT associated with cisplatin induced hearing loss in patients with cancer: Two new cohorts and a meta-analysis reveal significant heterogeneity between cohorts. PLoS ONE 2014, 9, e115869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.J.; Lim, J.Y.; Huang, J.; Bass, J.; Wu, J.; Wang, C.; Fang, J.; Stewart, E.; Harstead, E.H.; Robinson, G.W.; et al. The role of inherited TPMT and COMT genetic variation in cisplatin-induced ototoxicity in children with cancer. Clin. Pharmacol. Ther. 2013, 94, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Pussegoda, K.; Ross, C.J.; Visscher, H.; Yazdanpanah, M.; Brooks, B.; Rassekh, S.R.; Zada, Y.F.; Dube, M.P.; Carleton, B.C.; Hayden, M.R.; et al. Replication of TPMT and ABCC3 genetic variants highly associated with cisplatin-induced hearing loss in children. Clin. Pharmacol. Ther. 2013, 94, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y.G. Molecular basis of antifolate resistance. Cancer Metastasis Rev. 2007, 26, 153–181. [Google Scholar] [CrossRef] [PubMed]
- Farber, S.; Diamond, L.K. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Escherich, G.; Horstmann, M.A.; Zimmermann, M.; Janka-Schaub, G.E. Cooperative study group for childhood acute lymphoblastic leukaemia (COALL): Long-term results of trials 82, 85, 89, 92 and 97. Leukemia 2010, 24, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Lu, X.; Devidas, M.; Camitta, B.M.; Gaynon, P.S.; Winick, N.J.; Reaman, G.H.; Carroll, W.L. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: A report from the children’s oncology group. J. Clin. Oncol. 2012, 30, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Kodidela, S.; Suresh Chandra, P.; Dubashi, B. Pharmacogenetics of methotrexate in acute lymphoblastic leukaemia: Why still at the bench level? Eur. J. Clin. Pharmacol. 2014, 70, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Moricke, A.; Zimmermann, M.; Reiter, A.; Henze, G.; Schrauder, A.; Gadner, H.; Ludwig, W.D.; Ritter, J.; Harbott, J.; Mann, G.; et al. Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia 2010, 24, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, L.B.; Bruun, G.H.; Yang, W.; Trevino, L.R.; Vattathil, S.; Scheet, P.; Cheng, C.; Rosner, G.L.; Giacomini, K.M.; Fan, Y.; et al. Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 2012, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.; Evers, R.; Gupta, A.; Hop, C.E.; Salphati, L.; Shukla, S.; Ambudkar, S.V.; Unadkat, J.D. Interindividual variability in hepatic organic anion-transporting polypeptides and P-glycoprotein (ABCB1) protein expression: Quantification by liquid chromatography tandem mass spectroscopy and influence of genotype, age, and sex. Drug Metab. Dispos. 2014, 42, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Ulvestad, M.; Skottheim, I.B.; Jakobsen, G.S.; Bremer, S.; Molden, E.; Asberg, A.; Hjelmesaeth, J.; Andersson, T.B.; Sandbu, R.; Christensen, H. Impact of OATP1B1, MDR1, and CYP3A4 expression in liver and intestine on interpatient pharmacokinetic variability of atorvastatin in obese subjects. Clin. Pharmacol. Ther. 2013, 93, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.; Sauty, G.; Labuda, M.; Gagne, V.; Laverdiere, C.; Moghrabi, A.; Sinnett, D.; Krajinovic, M. Polymorphisms in multidrug resistance-associated protein gene 4 is associated with outcome in childhood acute lymphoblastic leukemia. Blood 2009, 114, 1383–1386. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.; Sauty, G.; Labuda, M.; Gagne, V.; Rousseau, J.; Moghrabi, A.; Laverdiere, C.; Sinnett, D.; Krajinovic, M. Polymorphism in multidrug resistance-associated protein gene 3 is associated with outcomes in childhood acute lymphoblastic leukemia. Pharmacogenom. J. 2012, 12, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Laverdiere, C.; Chiasson, S.; Costea, I.; Moghrabi, A.; Krajinovic, M. Polymorphism G80A in the reduced folate carrier gene and its relationship to methotrexate plasma levels and outcome of childhood acute lymphoblastic leukemia. Blood 2002, 100, 3832–3834. [Google Scholar] [CrossRef] [PubMed]
- Radtke, S.; Zolk, O.; Renner, B.; Paulides, M.; Zimmermann, M.; Moricke, A.; Stanulla, M.; Schrappe, M.; Langer, T. Germline genetic variations in methotrexate candidate genes are associated with pharmacokinetics, toxicity, and outcome in childhood acute lymphoblastic leukemia. Blood 2013, 121, 5145–5153. [Google Scholar] [CrossRef] [PubMed]
- Al-Shakfa, F.; Dulucq, S.; Brukner, I.; Milacic, I.; Ansari, M.; Beaulieu, P.; Moghrabi, A.; Laverdiere, C.; Sallan, S.E.; Silverman, L.B.; et al. DNA variants in region for noncoding interfering transcript of dihydrofolate reductase gene and outcome in childhood acute lymphoblastic leukemia. Clin. Cancer Res. 2009, 15, 6931–6938. [Google Scholar] [CrossRef] [PubMed]
- Dulucq, S.; St-Onge, G.; Gagne, V.; Ansari, M.; Sinnett, D.; Labuda, D.; Moghrabi, A.; Krajinovic, M. DNA variants in the dihydrofolate reductase gene and outcome in childhood ALL. Blood 2008, 111, 3692–3700. [Google Scholar] [CrossRef] [PubMed]
- Kodidela, S.; Pradhan, S.C.; Dubashi, B.; Basu, D. Influence of dihydrofolate reductase gene polymorphisms rs408626 (–317A>G) and rs442767 (–680C>A) on the outcome of methotrexate-based maintenance therapy in South Indian patients with acute lymphoblastic leukemia. Eur. J. Clin. Pharmacol. 2015, 71, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Krajinovic, M.; Costea, I.; Chiasson, S. Polymorphism of the thymidylate synthase gene and outcome of acute lymphoblastic leukaemia. Lancet 2002, 359, 1033–1034. [Google Scholar] [CrossRef]
- Krajinovic, M.; Costea, I.; Primeau, M.; Dulucq, S.; Moghrabi, A. Combining several polymorphisms of thymidylate synthase gene for pharmacogenetic analysis. Pharmacogenom. J. 2005, 5, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Relling, M.V.; Yang, W.; Das, S.; Cook, E.H.; Rosner, G.L.; Neel, M.; Howard, S.; Ribeiro, R.; Sandlund, J.T.; Pui, C.H.; et al. Pharmacogenetic risk factors for osteonecrosis of the hip among children with leukemia. J. Clin. Oncol. 2004, 22, 3930–3936. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.R.; Jones, G.M.; Narkewicz, M.R. Ontogeny of hepatic enzymes involved in serine- and folate-dependent one-carbon metabolism in rabbits. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G873–G878. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Thomas, D.; O'Brien, S.; Cortes, J.; Giles, F.; Jeha, S.; Bueso-Ramos, C.E.; Pierce, S.; Shan, J.; Koller, C.; et al. Long-term follow-up results of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (Hyper-CVAD), a dose-intensive regimen, in adult acute lymphocytic leukemia. Cancer 2004, 101, 2788–2801. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.A.; Dodge, R.K.; Burns, C.P.; Lee, E.J.; Stone, R.M.; Schulman, P.; Duggan, D.; Davey, F.R.; Sobol, R.E.; Frankel, S.R.; et al. A five-drug remission induction regimen with intensive consolidation for adults with acute lymphoblastic leukemia: Cancer and leukemia group B study 8811. Blood 1995, 85, 2025–2037. [Google Scholar] [PubMed]
- Gorlick, R.; Janeway, K.; Lessnick, S.; Randall, R.L.; Marina, N.; COG Bone Tumor Committee. Children’s Oncology Group’s 2013 blueprint for research: Bone tumors. Pediatr. Blood Cancer 2013, 60, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Malempati, S.; Hawkins, D.S. Rhabdomyosarcoma: Review of the Children’s Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr. Blood Cancer 2012, 59, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Bigner, S.H.; Bigner, D.D. Cyclophosphamide therapy of medulloblastoma: From the laboratory to the clinic and back again (and again and again). J. Neurooncol. 1995, 24, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tian, Q.; Yung Chan, S.; Chuen Li, S.; Zhou, S.; Duan, W.; Zhu, Y.Z. Metabolism and transport of oxazaphosphorines and the clinical implications. Drug Metab. Rev. 2005, 37, 611–703. [Google Scholar] [CrossRef] [PubMed]
- Bohnenstengel, F.; Hofmann, U.; Eichelbaum, M.; Kroemer, H.K. Characterization of the cytochrome P450 involved in side-chain oxidation of cyclophosphamide in humans. Eur. J. Clin. Pharmacol. 1996, 51, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.K.; Weber, G.F.; Crespi, C.L.; Waxman, D.J. Differential activation of cyclophosphamide and ifosphamide by cytochromes P-450 2B and 3A in human liver microsomes. Cancer Res. 1993, 53, 5629–5637. [Google Scholar] [PubMed]
- Ren, S.; Yang, J.S.; Kalhorn, T.F.; Slattery, J.T. Oxidation of cyclophosphamide to 4-hydroxycyclophosphamide and deschloroethylcyclophosphamide in human liver microsomes. Cancer Res. 1997, 57, 4229–4235. [Google Scholar] [PubMed]
- Moghe, A.; Ghare, S.; Lamoreau, B.; Mohammad, M.; Barve, S.; McClain, C.; Joshi-Barve, S. Molecular mechanisms of acrolein toxicity: Relevance to human disease. Toxicol. Sci. 2015, 143, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Dirven, H.A.; van Ommen, B.; van Bladeren, P.J. Involvement of human glutathione S-transferase isoenzymes in the conjugation of cyclophosphamide metabolites with glutathione. Cancer Res. 1994, 54, 6215–6120. [Google Scholar] [PubMed]
- Dirven, H.A.; Venekamp, J.C.; van Ommen, B.; van Bladeren, P.J. The interaction of glutathione with 4-hydroxycyclophosphamide and phosphoramide mustard, studied by 31P nuclear magnetic resonance spectroscopy. Chem. Biol. Interact. 1994, 93, 185–196. [Google Scholar] [CrossRef]
- Ekhart, C.; Doodeman, V.D.; Rodenhuis, S.; Smits, P.H.; Beijnen, J.H.; Huitema, A.D. Influence of polymorphisms of drug metabolizing enzymes (CYP2B6, CYP2C9, CYP2C19, CYP3A4, CYP3A5, GSTA1, GSTP1, ALDH1A1 and ALDH3A1) on the pharmacokinetics of cyclophosphamide and 4-hydroxycyclophosphamide. Pharmacogenet. Genom. 2008, 18, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Komagata, S.; Fujiki, Y.; Kanada, Y.; Ebi, H.; Itoh, K.; Mukai, H.; Yokoi, T.; Minami, H. Genetic polymorphisms of CYP2B6 affect the pharmacokinetics/pharmacodynamics of cyclophosphamide in Japanese cancer patients. Pharmacogenet. Genom. 2007, 17, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Timm, R.; Kaiser, R.; Lotsch, J.; Heider, U.; Sezer, O.; Weisz, K.; Montemurro, M.; Roots, I.; Cascorbi, I. Association of cyclophosphamide pharmacokinetics to polymorphic cytochrome P450 2C19. Pharmacogenom. J. 2005, 5, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Veal, G.J.; Cole, M.; Chinnaswamy, G.; Sludden, J.; Jamieson, D.; Errington, J.; Malik, G.; Hill, C.R.; Chamberlain, T.; Boddy, A.V. Cyclophosphamide pharmacokinetics and pharmacogenetics in children with B-cell non-Hodgkin’s lymphoma. Eur. J. Cancer 2016, 55, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Wagner, L.M. Fifteen years of irinotecan therapy for pediatric sarcoma: Where to next? Clin. Sarcoma Res. 2015, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Mathijssen, R.H.; van Alphen, R.J.; Verweij, J.; Loos, W.J.; Nooter, K.; Stoter, G.; Sparreboom, A. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin. Cancer Res. 2001, 7, 2182–2194. [Google Scholar] [PubMed]
- Saltz, L.B.; Cox, J.V.; Blanke, C.; Rosen, L.S.; Fehrenbacher, L.; Moore, M.J.; Maroun, J.A.; Ackland, S.P.; Locker, P.K.; Pirotta, N.; et al. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2000, 343, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Hines, R.N. The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol. Ther. 2008, 118, 250–267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, D.; Cui, D.; Gambardella, J.; Ma, L.; Barros, A.; Wang, L.; Fu, Y.; Rahematpura, S.; Nielsen, J.; et al. Characterization of the UDP glucuronosyltransferase activity of human liver microsomes genotyped for the UGT1A1*28 polymorphism. Drug Metab. Dispos. 2007, 35, 2270–2280. [Google Scholar] [CrossRef] [PubMed]
- Gagne, J.F.; Montminy, V.; Belanger, P.; Journault, K.; Gaucher, G.; Guillemette, C. Common human UGT1A polymorphisms and the altered metabolism of irinotecan active metabolite 7-ethyl-10-hydroxycamptothecin (SN-38). Mol. Pharmacol. 2002, 62, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Grimaldi, M.C.; Boyer, J.C.; Thomas, F.; Quaranta, S.; Picard, N.; Loriot, M.A.; Narjoz, C.; Poncet, D.; Gagnieu, M.C.; Ged, C.; et al. UGT1A1 genotype and irinotecan therapy: General review and implementation in routine practice. Fundam. Clin. Pharmacol. 2015, 29, 219–237. [Google Scholar] [CrossRef] [PubMed]
- DuBois, S.G.; Allen, S.; Bent, M.; Hilton, J.F.; Hollinger, F.; Hawkins, R.; Courtier, J.; Mosse, Y.P.; Matthay, K.K. Phase I/II study of 131I-MIBG with vincristine and 5 days of irinotecan for advanced neuroblastoma. Br. J. Cancer 2015, 112, 644–649. [Google Scholar] [CrossRef] [PubMed]
- DuBois, S.G.; Chesler, L.; Groshen, S.; Hawkins, R.; Goodarzian, F.; Shimada, H.; Yanik, G.; Tagen, M.; Stewart, C.; Mosse, Y.P.; et al. Phase I study of vincristine, irinotecan, and 131I-metaiodobenzylguanidine for patients with relapsed or refractory neuroblastoma: A new approaches to neuroblastoma therapy trial. Clin. Cancer Res. 2012, 18, 2679–2686. [Google Scholar] [CrossRef] [PubMed]
- Cole-Healy, Z.; Vergani, P.; Hunter, K.; Brown, N.J.; Reed, M.W.; Staton, C.A. The relationship between semaphorin 3C and microvessel density in the progression of breast and oral neoplasia. Exp. Mol. Pathol. 2015, 99, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Man, J.; Shoemake, J.; Zhou, W.; Fang, X.; Wu, Q.; Rizzo, A.; Prayson, R.; Bao, S.; Rich, J.N.; Yu, J.S. Sema3C promotes the survival and tumorigenicity of glioma stem cells through Rac1 activation. Cell. Rep. 2014, 9, 1812–1826. [Google Scholar] [CrossRef] [PubMed]
- Miyato, H.; Tsuno, N.H.; Kitayama, J. Semaphorin 3C is involved in the progression of gastric cancer. Cancer Sci. 2012, 103, 1961–1966. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Evans, W.E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 2006, 354, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Toso, R.J.; Thrower, D.; Wilson, L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc. Natl. Acad. Sci. USA 1993, 90, 9552–9556. [Google Scholar] [CrossRef] [PubMed]
- Egbelakin, A.; Ferguson, M.J.; MacGill, E.A.; Lehmann, A.S.; Topletz, A.R.; Quinney, S.K.; Li, L.; McCammack, K.C.; Hall, S.D.; Renbarger, J.L. Increased risk of vincristine neurotoxicity associated with low CYP3A5 expression genotype in children with acute lymphoblastic leukemia. Pediatr. Blood Cancer 2011, 56, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.G.; Wood, A.J.; Kim, R.B.; Stein, C.M.; Wilkinson, G.R. Genetic variability in CYP3A5 and its possible consequences. Pharmacogenomics 2004, 5, 243–272. [Google Scholar] [CrossRef] [PubMed]
- Dennison, J.B.; Jones, D.R.; Renbarger, J.L.; Hall, S.D. Effect of CYP3A5 expression on vincristine metabolism with human liver microsomes. J. Pharmacol. Exp. Ther. 2007, 321, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Dennison, J.B.; Kulanthaivel, P.; Barbuch, R.J.; Renbarger, J.L.; Ehlhardt, W.J.; Hall, S.D. Selective metabolism of vincristine in vitro by CYP3A5. Drug Metab. Dispos. 2006, 34, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Guilhaumou, R.; Simon, N.; Quaranta, S.; Verschuur, A.; Lacarelle, B.; Andre, N.; Solas, C. Population pharmacokinetics and pharmacogenetics of vincristine in paediatric patients treated for solid tumour diseases. Cancer Chemother. Pharmacol. 2011, 68, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Norris, R.; Price, G.; Nguyen, T.; Ni, M.; George, R.; van Breda, K.; Duley, J.; Charles, B.; Pinkerton, R. Vincristine pharmacodynamics and pharmacogenetics in children with cancer: A limited-sampling, population modelling approach. J. Paediatr. Child. Health 2011, 47, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.P. The effect of race on the CYP3A-mediated metabolism of vincristine in pediatric patients with acute lymphoblastic leukemia. J. Oncol. Pharm. Pract. 2016, 22, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Diouf, B.; Crews, K.R.; Lew, G.; Pei, D.; Cheng, C.; Bao, J.; Zheng, J.J.; Yang, W.; Fan, Y.; Wheeler, H.E.; et al. Association of an inherited genetic variant with vincristine-related peripheral neuropathy in children with acute lymphoblastic leukemia. JAMA 2015, 313, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Camino, A.; Martin-Guerrero, I.; Lopez-Lopez, E.; Echebarria-Barona, A.; Zabalza, I.; Ruiz, I.; Guerra-Merino, I.; Garcia-Orad, A. Lack of association of the CEP72 rs924607 TT genotype with vincristine-related peripheral neuropathy during the early phase of pediatric acute lymphoblastic leukemia treatment in a Spanish population. Pharmacogenet. Genom. 2016, 26, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Bosilkovska, M.; Ing Lorenzini, K.; Uppugunduri, C.R.; Desmeules, J.; Daali, Y.; Escher, M. Severe Vincristine-induced Neuropathic Pain in a CYP3A5 Nonexpressor With Reduced CYP3A4/5 Activity: Case Study. Clin. Ther. 2016, 38, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Ceppi, F.; Langlois-Pelletier, C.; Gagne, V.; Rousseau, J.; Ciolino, C.; de Lorenzo, S.; Kevin, K.M.; Cijov, D.; Sallan, S.E.; Silverman, L.B.; et al. Polymorphisms of the vincristine pathway and response to treatment in children with childhood acute lymphoblastic leukemia. Pharmacogenomics 2014, 15, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Sauerbrey, A.; Steinbach, D.; Gebhart, E.; Drexler, H.G.; Miyachi, H.; Chitambar, C.R.; Becker, C.M.; Zintl, F.; Humeny, A. Analysis of single nucleotide polymorphism C3435T of the multidrug resistance gene MDR1 in acute lymphoblastic leukemia. Int. J. Oncol. 2003, 23, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Jamroziak, K.; Balcerczak, E.; Cebula, B.; Kowalczyk, M.; Panczyk, M.; Janus, A.; Smolewski, P.; Mirowski, M.; Robak, T. Multi-drug transporter MDR1 gene polymorphism and prognosis in adult acute lymphoblastic leukemia. Pharmacol. Rep. 2005, 57, 882–888. [Google Scholar] [PubMed]
- Yang, J.J.; Cheng, C.; Devidas, M.; Cao, X.; Campana, D.; Yang, W.; Fan, Y.; Neale, G.; Cox, N.; Scheet, P.; et al. Genome-wide association study identifies germline polymorphisms associated with relapse of childhood acute lymphoblastic leukemia. Blood 2012, 120, 4197–4204. [Google Scholar] [CrossRef] [PubMed]






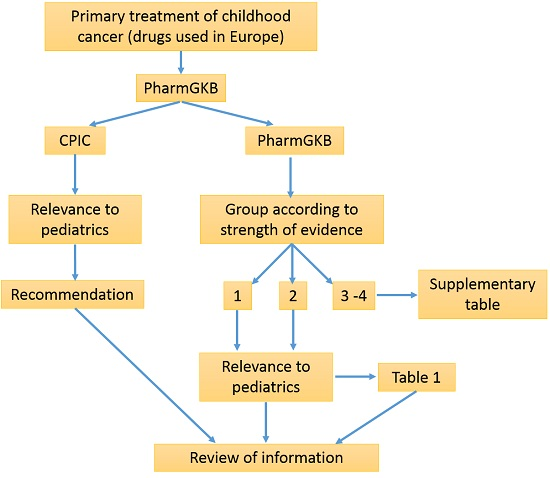{kind=link}
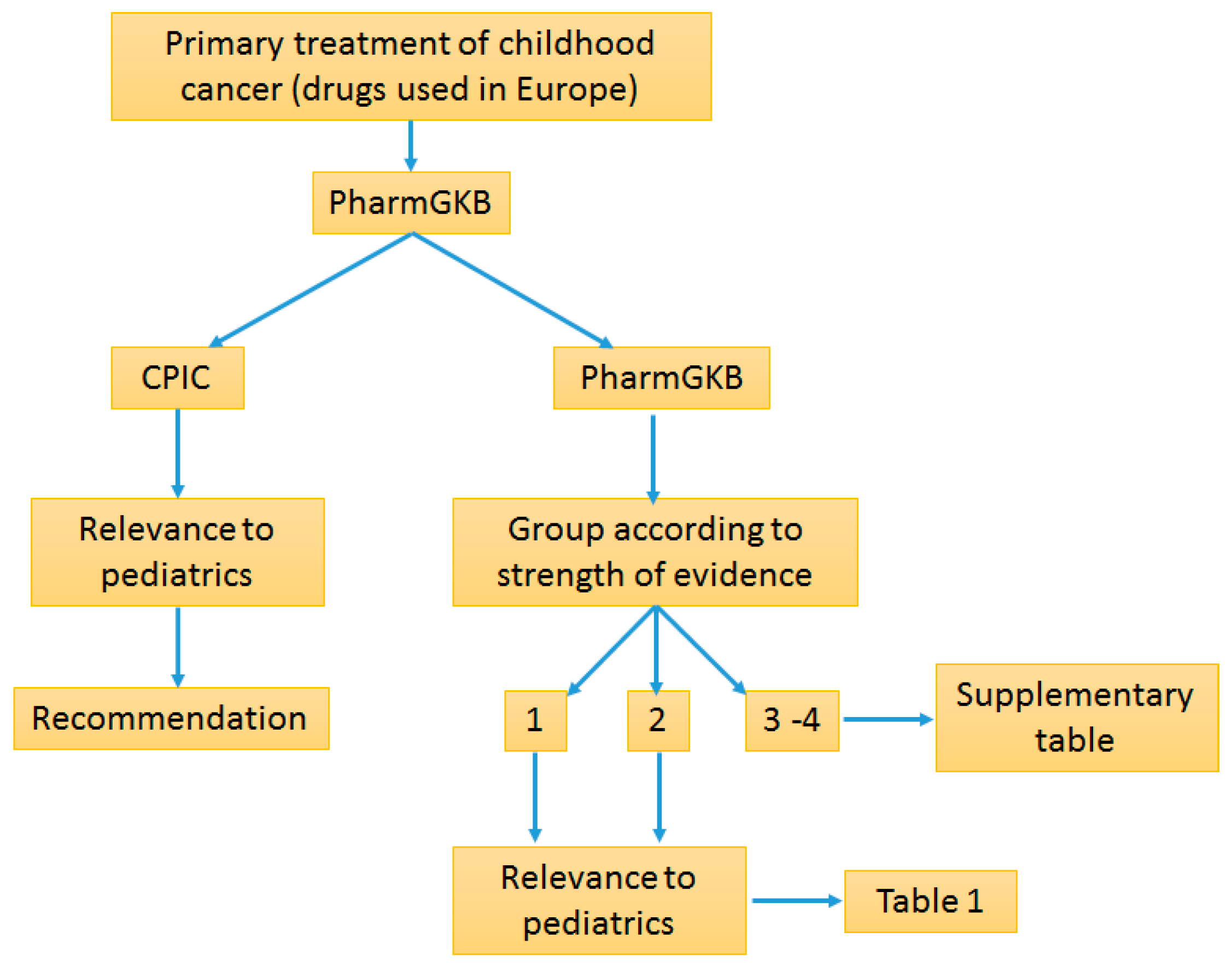{kind=link}
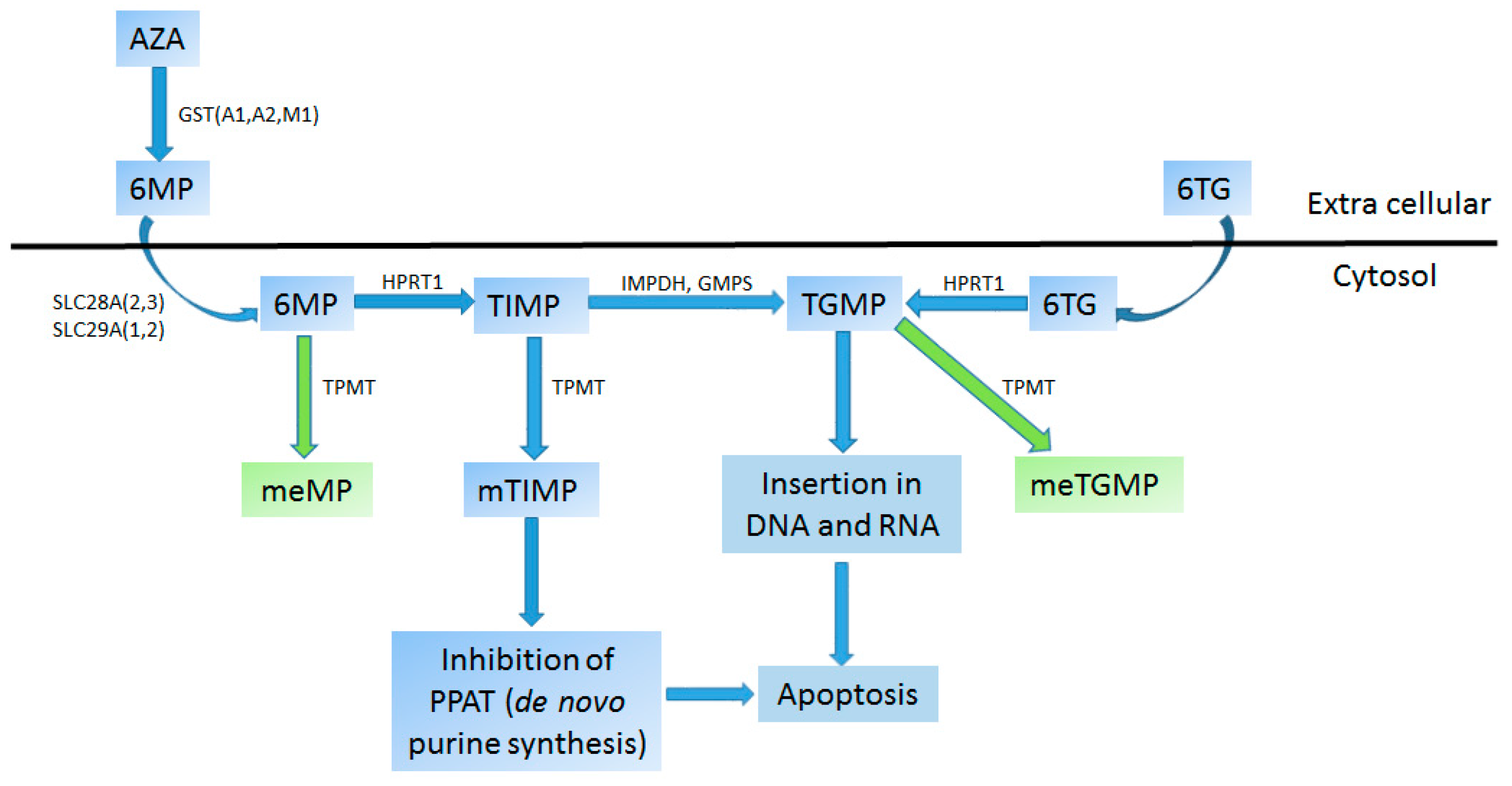{kind=link}
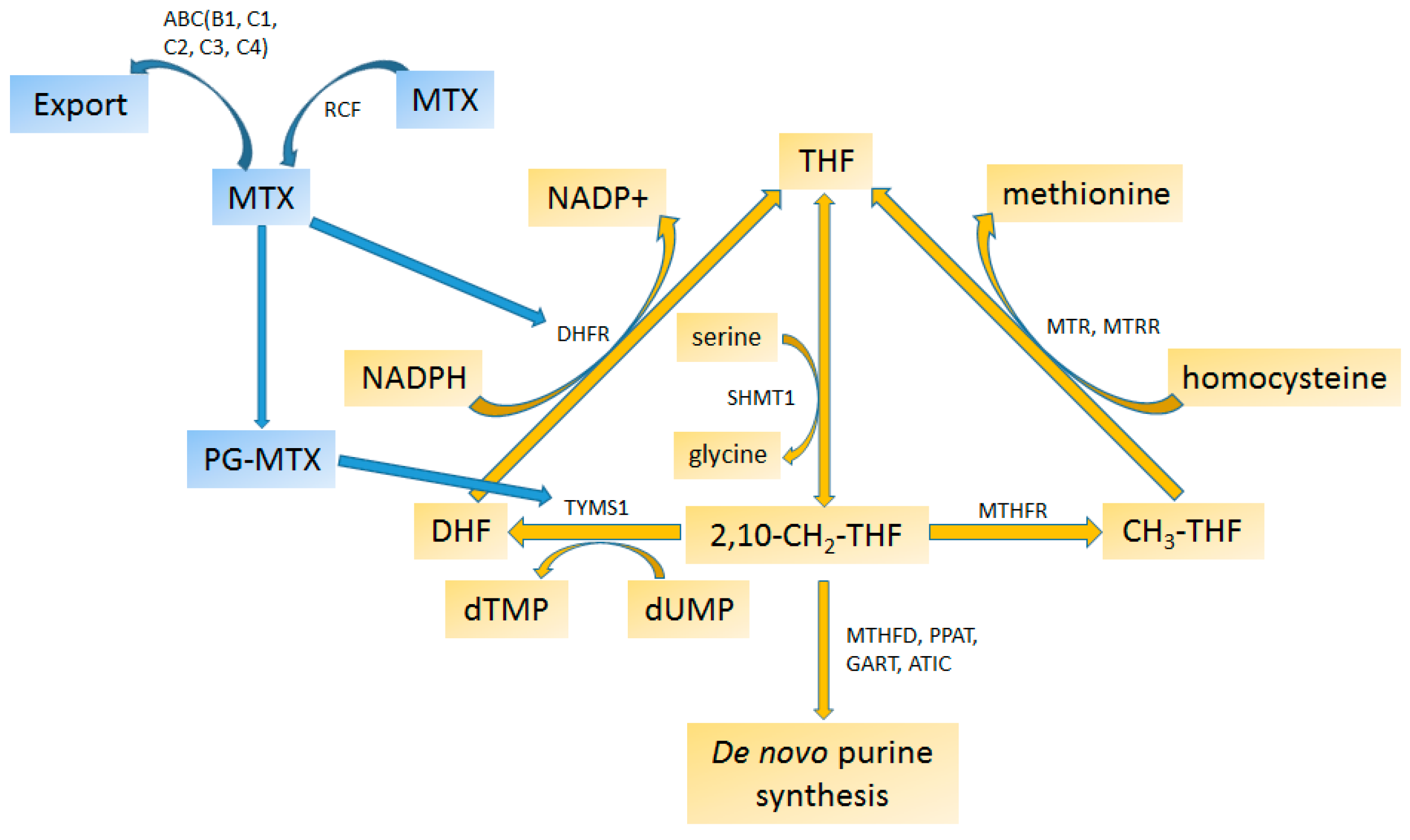{kind=link}
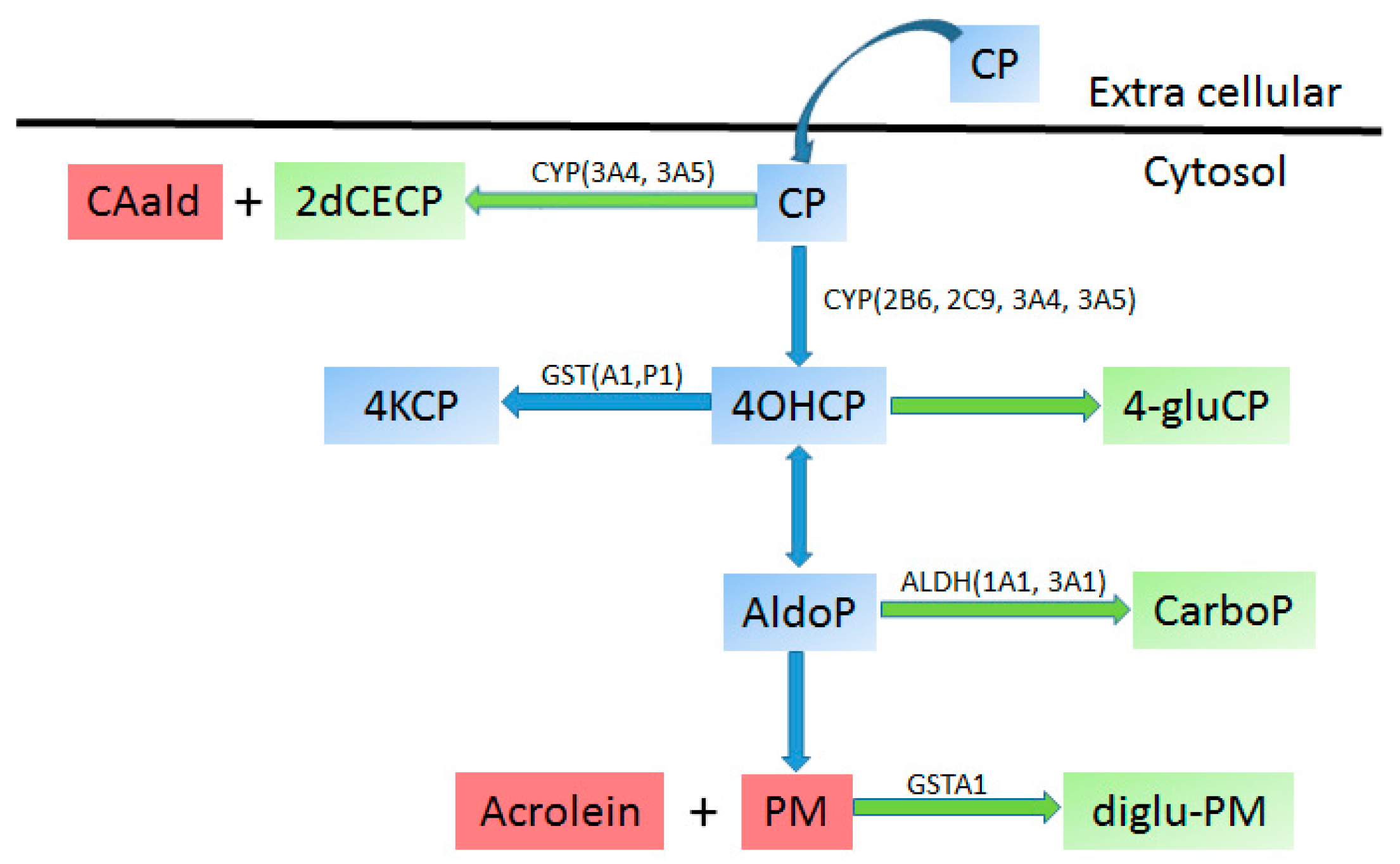{kind=link}
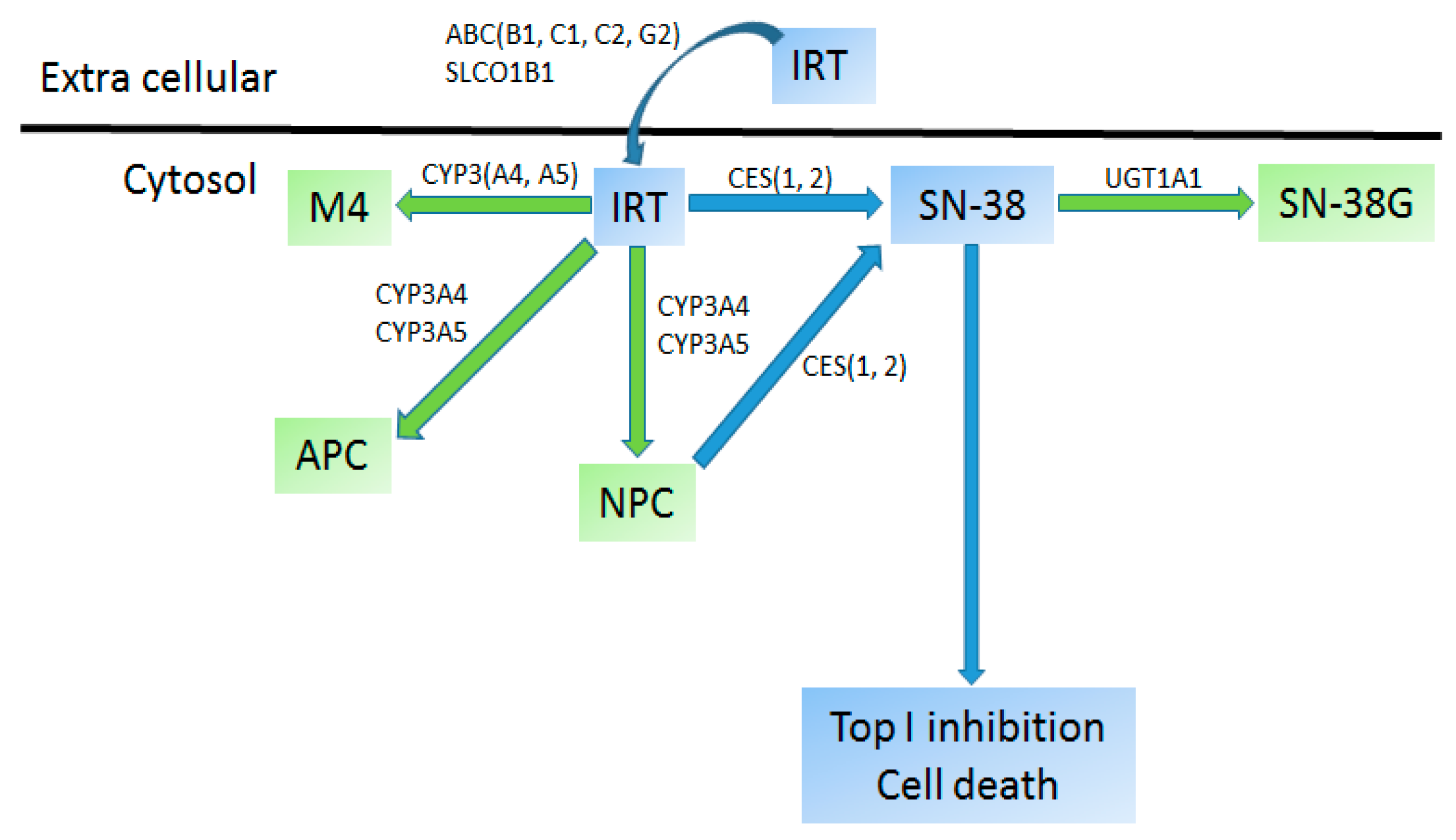{kind=link}
| Evidence Level | Drug | Gene | Allele/Variant | AFR | EAS | EUR | Effect | Total Articles | Pediatric Articles | All ref. | Ped. ref. | Condition (Pediatric) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Level 1 | Thiopurines | TPMT | *2 rs1800462 | 0.001 | 0.000 | 0.006 | Dosage, Toxicity/ADR | 96 | 30 | [23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119] | [89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118] | Acute lymphoblastic leukemia |
| *3B rs1800460 | 0.003 | 0.000 | 0.028 | |||||||||
| *3C rs1142345 | 0.067 | 0.022 | 0.029 | |||||||||
| *4 rs1800584 | NA | NA | NA | |||||||||
| NUDT15 | *2 and *3 rs116855232 | 0.008 | 0.095 | 0.002 | Dosage, Toxicity/ADR | 9 | 6 | [120,121,122,123,124,125,126,127,128,129,130,131,132] | [120,121,122,123,124,125,126] | Acute lymphoblastic leukemia | ||
| Cisplatin | XPC | rs2228001 | 0.249 | 0.333 | 0.405 | Toxicity/ADR | 2 | 1 | [133,134] | [133] | Osteosarcoma | |
| Level 2 | Cisplatin | ERCC1 | rs3212986 rs11615 | 0.291 0.037 | 0.299 0.262 | 0.250 0.622 | Efficacy, Toxicity/ADR | 11 | 0 | [135,136,137,138,139,140,141,142,143,144,145,146,147] | NS | |
| GSTM1 | Null | NA | NA | NA | Efficacy | 3 | 0 | [148,149,150] | NS | |||
| TP53 | rs1042522 | 0.669 | 0.414 | 0.285 | Efficacy, Toxicity/ADR | 5 | 0 | [139,140,151,152,153] | NS | |||
| XRCC1 | rs25487 | 0.110 | 0.235 | 0.366 | Toxicity/ADR | 9 | 0 | [136,137,138,139,140,154,155,156,157] | NS | |||
| Carboplatin | EGFR | rs121434568 | NA | NA | NA | Efficacy | 8 | 0 | [158,159,160,161,162,163,164,165] | NS | ||
| ERCC1 | rs11615 | 0.037 | 0.262 | 0.622 | Efficacy, Toxicity/ADR | 11 | 0 | [135,136,137,138,139,140,141,142,143,144,145,146,147] | NS | |||
| MTHFR | rs1801133 | 0.090 | 0.296 | 0.365 | Efficacy | 2 | 0 | [166,167] | NS | |||
| XRCC1 | rs25487 | 0.110 | 0.235 | 0.366 | Efficacy, Toxicity/ADR | 9 | 0 | [136,137,138,139,140,154,155,156,157] | NS | |||
| Methotrexate | ABCB1 | rs1045642 | 0.150 | 0.398 | 0.518 | Toxicity/ADR | 3 | 2 | [168,169,170] | [168,170] | Lymphoma | |
| ATIC | rs4673993 | 0.095 | 0.294 | 0.313 | Efficacy | 2 | 0 | [171,172] | NS | |||
| MTHFR | rs1801133 | 0.090 | 0.295 | 0.365 | Efficacy, Toxicity/ADR | 37 | 26 | [100,169,170,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206] | [100,170,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196] | Acute lymphoblastic leukemia | ||
| MTRR | rs1801394 | 0.246 | 0.263 | 0.523 | Toxicity/ADR, Metabolism/PK | 3 | 3 | [185,207,208] | [185,207,208] | Acute lymphoblastic leukemia | ||
| SLCO1B1 | rs11045879 | 0.189 | 0.453 | 0.190 | Toxicity/ADR | 4 | 3 | [209,210,211,212] | [209,210,211] | Acute lymphoblastic leukemia | ||
| Level 2 | Cyclophosphamide | GSTP1 | rs1695 | 0.480 | 0.179 | 0.331 | Efficacy, Toxicity/ADR | 2 | 0 | [213,214] | NS | |
| MTHFR | rs1801133 | 0.090 | 0.296 | 0.365 | Toxicity/ADR | 3 | 1 | [151,188,204] | [188] | Osteosarcoma | ||
| SOD2 | rs4880 | 0.424 | 0.125 | 0.466 | Efficacy | 1 | 0 | [215] | NS | |||
| TP53 | rs1042522 | 0.669 | 0.414 | 0.285 | Efficacy, Toxicity/ADR | 5 | 0 | [139,140,151,152,153] | NS | |||
| Irinotecan | C8orf34 | rs1517114 | 0.424 | 0.122 | 0.363 | Toxicity/ADR | 1 | 0 | [216] | NS | ||
| SEMA3C | rs7779029 | 0.365 | 0.152 | 0.047 | Toxicity/ADR | 1 | 0 | [216] | NS | |||
| UGT1A1 | rs8175347 rs4148323 | NA 0.001 | NA 0.138 | NA 0.007 | Toxicity/ADR | 35 | 1 | [217,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248,249,250,251,252,253,254] | [249] | Solid tumors |
| TPMT Phenotype/Genotype | Dosing Recommendation 6MP | Dosing Recommendation 6TG |
|---|---|---|
| Normal metabolizer (two functional alleles) | Start with normal dose | Start with normal dose |
| Intermediate metabolizer (one functional allele) | Start with 30% to 70% reduced dose | Start with 30% to 50% reduced dose |
| Poor metabolizer (no functional alleles) | Start with 90% reduced dose, trice weekly | Start with 90% reduced dose, trice weekly |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mlakar, V.; Huezo-Diaz Curtis, P.; Satyanarayana Uppugunduri, C.R.; Krajinovic, M.; Ansari, M. Pharmacogenomics in Pediatric Oncology: Review of Gene—Drug Associations for Clinical Use. Int. J. Mol. Sci. 2016, 17, 1502. https://doi.org/10.3390/ijms17091502
Mlakar V, Huezo-Diaz Curtis P, Satyanarayana Uppugunduri CR, Krajinovic M, Ansari M. Pharmacogenomics in Pediatric Oncology: Review of Gene—Drug Associations for Clinical Use. International Journal of Molecular Sciences. 2016; 17(9):1502. https://doi.org/10.3390/ijms17091502
Chicago/Turabian StyleMlakar, Vid, Patricia Huezo-Diaz Curtis, Chakradhara Rao Satyanarayana Uppugunduri, Maja Krajinovic, and Marc Ansari. 2016. "Pharmacogenomics in Pediatric Oncology: Review of Gene—Drug Associations for Clinical Use" International Journal of Molecular Sciences 17, no. 9: 1502. https://doi.org/10.3390/ijms17091502
APA StyleMlakar, V., Huezo-Diaz Curtis, P., Satyanarayana Uppugunduri, C. R., Krajinovic, M., & Ansari, M. (2016). Pharmacogenomics in Pediatric Oncology: Review of Gene—Drug Associations for Clinical Use. International Journal of Molecular Sciences, 17(9), 1502. https://doi.org/10.3390/ijms17091502