
Ion Channels in Brain Metastasis


Abstract
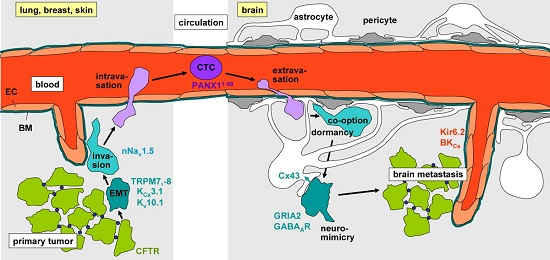
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
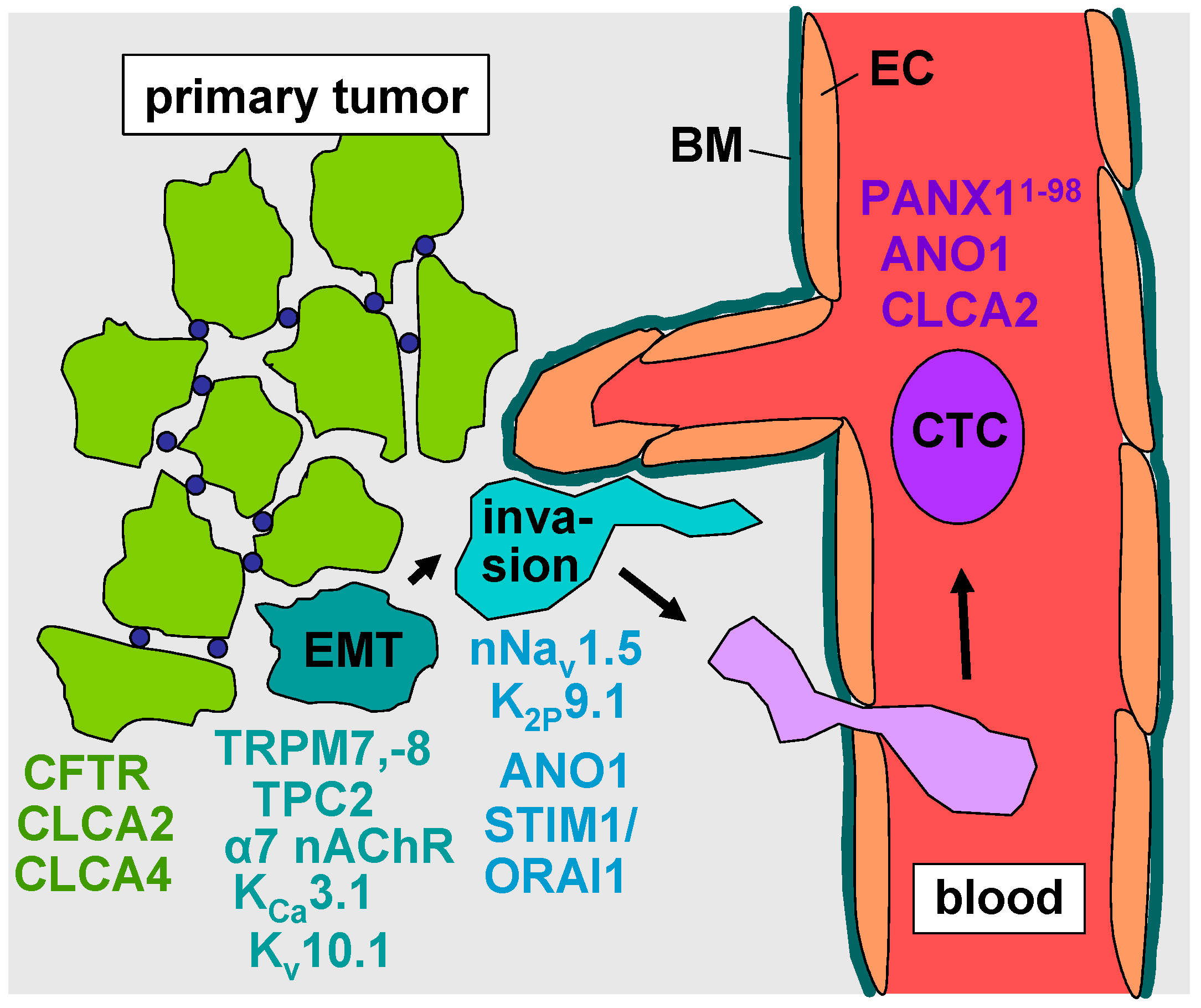
2. Epithelial–Mesenchymal Transition
3. Evasion from the Primary Tumor
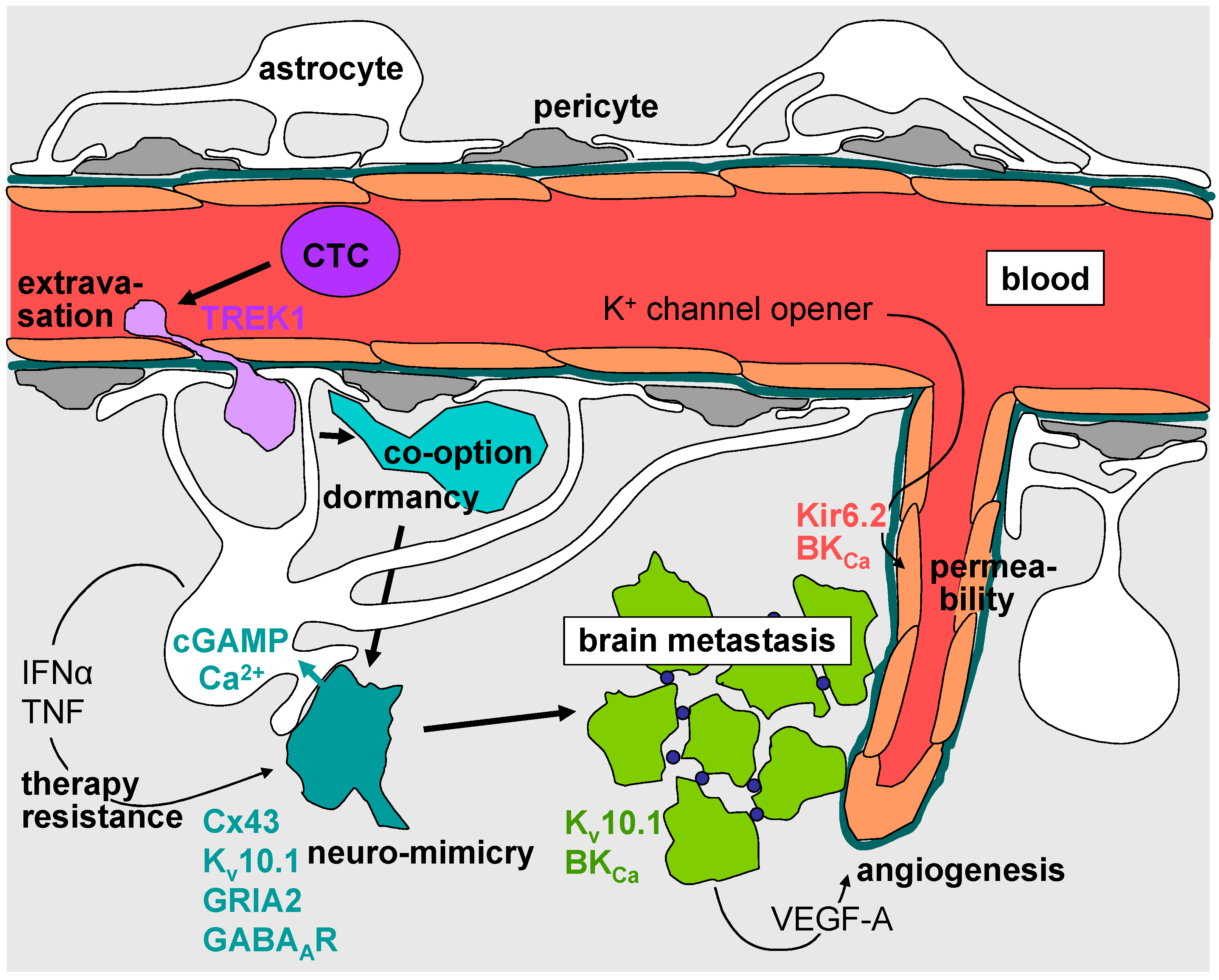
4. Intravasation, Circulation, Brain Tropism, and Transmigration of the Blood–Brain Barrier
5. Formation of the Metastatic Niche, Neuro-Mimicry, Brain Colonization, and Angiogenesis
6. Radiotherapy of Brain Metastases
7. Targeting of Brain Metastasis-Promoting Ion Channels
8. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- German-Cancer-Society. Available online: https://www.krebsgesellschaft.de/onko-internetportal/basis-informationen-krebs/krebsarten/hirntumor/hirnm (accessed on 15 July 2016).
- Weidle, U.H.; Birzele, F.; Kollmorgen, G.; Ruger, R. Dissection of the process of brain metastasis reveals targets and mechanisms for molecular-based intervention. Cancer Genom. Proteom. 2016, 13, 245–258. [Google Scholar]
- Kamp, M.A.; Slotty, P.J.; Cornelius, J.F.; Steiger, H.J.; Rapp, M.; Sabel, M. The impact of cerebral metastases growth pattern on neurosurgical treatment. Neurosurg. Rev. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lee, H.W.; Kim, Y.; Lee, Y.; Choi, Y.S.; Kim, K.H.; Jin, J.; Lee, J.; Joo, K.M.; Nam, D.H. Radiosensitization of brain metastasis by targeting c-MET. Lab. Investig. 2013, 93, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.S.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.M. Oncochannels. Cell Calcium 2013, 53, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.M.; Butz, L.; Stegen, B.; Klumpp, D.; Braun, N.; Ruth, P.; Eckert, F. Ionizing radiation, ion transports, and radioresistance of cancer cells. Front. Physiol. 2013, 4, 212. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.M.; Butz, L.; Stegen, B.; Klumpp, L.; Klumpp, D.; Eckert, F. Role of ion channels in ionizing radiation-induced cell death. Biochim. Biophys. Acta 2015, 1848, 2657–2664. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, A.; Hofmann, G.; Pillozzi, S.; Guasti, L.; Crociani, O.; Cilia, E.; Di Stefano, P.; Degani, S.; Balzi, M.; Olivotto, M.; et al. Human ether-a-go-go-related gene 1 channels are physically linked to β1 integrins and modulate adhesion-dependent signaling. Mol. Biol. Cell 2005, 16, 2972–2983. [Google Scholar] [CrossRef] [PubMed]
- Pardo, L.A.; del Camino, D.; Sanchez, A.; Alves, F.; Bruggemann, A.; Beckh, S.; Stühmer, W. Oncogenic potential of EAG K+ channels. EMBO J. 1999, 18, 5540–5547. [Google Scholar] [CrossRef] [PubMed]
- Stemmler, H.J.; Schmitt, M.; Willems, A.; Bernhard, H.; Harbeck, N.; Heinemann, V. Ratio of trastuzumab levels in serum and cerebrospinal fluid is altered in HER2-positive breast cancer patients with brain metastases and impairment of blood-brain barrier. Anticancer Drugs 2007, 18, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Svokos, K.A.; Salhia, B.; Toms, S.A. Molecular biology of brain metastasis. Int. J. Mol. Sci. 2014, 5, 9519–9530. [Google Scholar] [CrossRef] [PubMed]
- Wazny, L.D.; Brophy, D.F. Amiloride for the prevention of amphotericin B-induced hypokalemia and hypomagnesemia. Ann. Pharmacother. 2000, 34, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Bagal, S.K.; Brown, A.D.; Cox, P.J.; Omoto, K.; Owen, R.M.; Pryde, D.C.; Sidders, B.; Skerratt, S.E.; Stevens, E.B.; Storer, R.I.; et al. Ion channels as therapeutic targets: A drug discovery perspective. J. Med. Chem. 2013, 56, 593–624. [Google Scholar] [CrossRef] [PubMed]
- Keune, P.M.; Cocks, A.J.; Young, W.R.; Burschka, J.M.; Hansen, S.; Hofstadt-van Oy, U.; Oschmann, P.; Muenssinger, J. Dynamic walking features and improved walking performance in multiple sclerosis patients treated with fampridine (4-aminopyridine). BMC Neurol. 2015, 15, 171. [Google Scholar] [CrossRef] [PubMed]
- Cholon, D.M.; Esther, C.R., Jr.; Gentzsch, M. Efficacy of lumacaftor-ivacaftor for the treatment of cystic fibrosis patients homozygous for the F508del-CFTR mutation. Expert Rev. Precis. Med. Drug Dev. 2016, 1, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Ataga, K.I.; Smith, W.R.; De Castro, L.M.; Swerdlow, P.; Saunthararajah, Y.; Castro, O.; Vichinsky, E.; Kutlar, A.; Orringer, E.P.; Rigdon, G.C.; et al. Efficacy and safety of the Gardos channel blocker, senicapoc (ICA-17043), in patients with sickle cell anemia. Blood 2008, 111, 3991–3997. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Manago, A.; Zoratti, M.; Gulbins, E.; Szabo, I. Pharmacological targeting of ion channels for cancer therapy: In vivo evidences. Biochim. Biophys. Acta 2016, 1863, 1385–1397. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Qin, K.; Zhang, Y.; Gong, J.; Li, N.; Lv, D.; Xiang, R.; Tan, X. Downregulation of transcription factor Oct4 induces an epithelial-to-mesenchymal transition via enhancement of Ca2+ influx in breast cancer cells. Biochem. Biophys. Res. Commun. 2011, 411, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Peters, A.A.; Grice, D.M.; Cabot, P.J.; Parat, M.O.; Roberts-Thomson, S.J.; Monteith, G.R. Non-stimulated, agonist-stimulated and store-operated Ca2+ influx in MDA-MB-468 breast cancer cells and the effect of EGF-induced EMT on calcium entry. PLoS ONE 2012, 7, e36923. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, S.H.; Cheng, H.; Li, J.; Feng, R. The effect of TGF-β-induced epithelial-mesenchymal transition on the expression of intracellular calcium-handling proteins in T47D and MCF-7 human breast cancer cells. Arch. Biochem. Biophys. 2015, 583, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Azimi, I.; Faville, R.A.; Peters, A.A.; Jalink, K.; Putney, J.W., Jr.; Goodhill, G.J.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene 2014, 33, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, Y.; Shuai, S.; Ding, D.; Li, R.; Luo, R. TRPM8 promotes aggressiveness of breast cancer cells by regulating EMT via activating AKT/GSK-3β pathway. Tumour Biol. 2014, 35, 8969–8977. [Google Scholar] [CrossRef] [PubMed]
- Jahidin, A.H.; Stewart, T.A.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Differential effects of two-pore channel protein 1 and 2 silencing in MDA-MB-468 breast cancer cells. Biochem. Biophys. Res. Commun. 2016, 477, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, P.; Rizwani, W.; Pillai, S.; Kinkade, R.; Kovacs, M.; Rastogi, S.; Banerjee, S.; Carless, M.; Kim, E.; Coppola, D.; et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int. J. Cancer 2009, 124, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Restrepo-Angulo, I.; Sanchez-Torres, C.; Camacho, J. Human EAG1 potassium channels in the epithelial-to-mesenchymal transition in lung cancer cells. Anticancer Res. 2011, 31, 1265–1270. [Google Scholar] [PubMed]
- Arthur, G.K.; Duffy, S.M.; Roach, K.M.; Hirst, R.A.; Shikotra, A.; Gaillard, E.A.; Bradding, P. KCa3.1 K+Channel expression and function in human bronchial epithelial cells. PLoS ONE 2015, 10, e0145259. [Google Scholar] [CrossRef] [PubMed]
- Ramena, G.; Yin, Y.; Yu, Y.; Walia, V.; Elble, R.C. CLCA2 Interactor EVA1 is required for mammary epithelial cell differentiation. PLoS ONE 2016, 11, e0147489. [Google Scholar] [CrossRef] [PubMed]
- Walia, V.; Yu, Y.; Cao, D.; Sun, M.; McLean, J.R.; Hollier, B.G.; Cheng, J.; Mani, S.A.; Rao, K.; Premkumar, L.; et al. Loss of breast epithelial marker hCLCA2 promotes epithelial-to-mesenchymal transition and indicates higher risk of metastasis. Oncogene 2013, 31, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Walia, V.; Elble, R.C. Loss of CLCA4 promotes epithelial-to-mesenchymal transition in breast cancer cells. PLoS ONE 2013, 8, e83943. [Google Scholar] [CrossRef] [PubMed]
- Fuller, C.M.; Benos, D.J. Electrophysiological characteristics of the Ca2+-activated Cl− channel family of anion transport proteins. Clin. Exp. Pharm. Physiol. 2000, 27, 906–910. [Google Scholar] [CrossRef]
- Elble, R.C.; Walia, V.; Cheng, H.C.; Connon, C.J.; Mundhenk, L.; Gruber, A.D.; Pauli, B.U. The putative chloride channel hCLCA2 has a single C-terminal transmembrane segment. J. Biol. Chem. 2006, 281, 29448–29454. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Koyama, R.; Maruyama, R.; Hirano, T.; Tamura, M.; Sugisaka, J.; Suzuki, H.; Idogawa, M.; Shinomura, Y.; Tokino, T. CLCA2, a target of the p53 family, negatively regulates cancer cell migration and invasion. Cancer Biol. Ther. 2012, 13, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cowell, J.K.; Sossey-Alaoui, K. CLCA2 tumour suppressor gene in 1p31 is epigenetically regulated in breast cancer. Oncogene 2004, 23, 1474–1480. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.T.; Jiang, X.H.; Xie, C.; Cheng, H.; Da Dong, J.; Wang, Y.; Fok, K.L.; Zhang, X.H.; Sun, T.T.; Tsang, L.L.; et al. Downregulation of CFTR promotes epithelial-to-mesenchymal transition and is associated with poor prognosis of breast cancer. Biochim. Biophys. Acta 2013, 1833, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Brackenbury, W.J. Voltage-gated sodium channels and metastatic disease. Channels 2012, 6, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Fraser, S.P.; Ozerlat-Gunduz, I.; Brackenbury, W.J.; Fitzgerald, E.M.; Campbell, T.M.; Coombes, R.C.; Djamgoz, M.B. Regulation of voltage-gated sodium channel expression in cancer: Hormones, growth factors and auto-regulation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130105. [Google Scholar] [CrossRef] [PubMed]
- Campbell, T.M.; Main, M.J.; Fitzgerald, E.M. Functional expression of the voltage-gated Na+ channel Nav1.7 is necessary for EGF-mediated invasion in human non-small cell lung cancer cells. J. Cell Sci. 2013, 26, 4939–4949. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.; Millican-Slater, R.; Forrest, L.C.; Brackenbury, W.J. The sodium channel β1 subunit mediates outgrowth of neurite-like processes on breast cancer cells and promotes tumour growth and metastasis. Int. J. Cancer 2014, 135, 2338–2351. [Google Scholar] [CrossRef] [PubMed]
- Fraser, S.P.; Diss, J.K.; Chioni, A.M.; Mycielska, M.E.; Pan, H.; Yamaci, R.F.; Pani, F.; Siwy, Z.; Krasowska, M.; Grzywna, Z.; et al. Voltage-gated sodium channel expression and potentiation of human breast cancer metastasis. Clin. Cancer Res. 2005, 11, 5381–5389. [Google Scholar] [CrossRef] [PubMed]
- Brackenbury, W.J.; Chioni, A.M.; Diss, J.K.; Djamgoz, M.B. The neonatal splice variant of Nav1.5 potentiates in vitro invasive behaviour of MDA-MB-231 human breast cancer cells. Breast Cancer Res. Treat. 2007, 101, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Kozminski, D.J.; Wold, L.A.; Modak, R.; Calhoun, J.D.; Isom, L.L.; Brackenbury, W.J. Therapeutic potential for phenytoin: Targeting Nav1.5 sodium channels to reduce migration and invasion in metastatic breast cancer. Breast Cancer Res. Treat. 2012, 134, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.; Yang, M.; Millican-Slater, R.; Brackenbury, W.J. Nav1.5 regulates breast tumor growth and metastatic dissemination in vivo. Oncotarget 2015, 6, 32914–32929. [Google Scholar] [PubMed]
- Nelson, M.; Yang, M.; Dowle, A.A.; Thomas, J.R.; Brackenbury, W.J. The sodium channel-blocking antiepileptic drug phenytoin inhibits breast tumour growth and metastasis. Mol. Cancer 2015, 14, 13. [Google Scholar] [CrossRef] [PubMed]
- Driffort, V.; Gillet, L.; Bon, E.; Marionneau-Lambot, S.; Oullier, T.; Joulin, V.; Collin, C.; Pagès, J.C.; Jourdan, M.L.; Chevalier, S.; et al. Ranolazine inhibits Nav1.5-mediated breast cancer cell invasiveness and lung colonization. Mol. Cancer 2014, 13, 264. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, S.; Altun, S.; Gumushan, H.; Patel, A.; Djamgoz, M.B. Voltage-gated sodium channel activity promotes prostate cancer metastasis in vivo. Cancer Lett. 2012, 323, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, F.H.; Khajah, M.A.; Yang, M.; Brackenbury, W.J.; Luqmani, Y.A. Blockade of voltage-gated sodium channels inhibits invasion of endocrine-resistant breast cancer cells. Int. J. Oncol. 2016, 48, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.; Roger, S.; Besson, P.; Lecaille, F.; Gore, J.; Bougnoux, P.; Lalmanach, G.; Le Guennec, J.Y. Voltage-gated sodium channel activity promotes cysteine cathepsin-dependent invasiveness and colony growth of human cancer cells. J. Biol. Chem. 2009, 284, 8680–8691. [Google Scholar] [CrossRef] [PubMed]
- Brisson, L.; Gillet, L.; Calaghan, S.; Besson, P.; le Guennec, J.Y.; Roger, S.; Gore, J. Nav1.5 enhances breast cancer cell invasiveness by increasing NHE1-dependent H+ efflux in caveolae. Oncogene 2011, 30, 2070–2076. [Google Scholar] [CrossRef] [PubMed]
- Carrithers, M.D.; Chatterjee, G.; Carrithers, L.M.; Offoha, R.; Iheagwara, U.; Rahner, C.; Graham, M.; Waxman, S.G. Regulation of podosome formation in macrophages by a splice variant of the sodium channel SCN8A. J. Biol. Chem. 2009, 284, 8114–8126. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Shang, L.Q.; Chen, R.L.; Yang, S.M.; Wang, S.L.; Wang, J.; Sun, G. Significance of Trask protein interactions in brain metastatic cohorts of lung cancers. Tumour Biol. 2015, 36, 4181–4187. [Google Scholar] [CrossRef] [PubMed]
- Didiasova, M.; Zakrzewicz, D.; Magdolen, V.; Nagaraj, C.; Balint, Z.; Rohde, M.; Preissner, K.T.; Wygrecka, M. STIM1/ORAI1-mediated Ca2+ influx regulates enolase-1 exteriorization. J. Biol. Chem. 2015, 290, 11983–11999. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Luo, L.; Lal, B.; Ma, X.; Chen, L.; Hann, C.L.; Fulton, A.M.; Leahy, D.J.; Laterra, J.; Li, M. A monoclonal antibody against KCNK9 K+ channel extracellular domain inhibits tumour growth and metastasis. Nat. Commun. 2016, 7, 10339. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Liu, W.; Guan, L.; Lu, M.; Wang, K. Inhibition of Calcium-Activated Chloride Channel ANO1/TMEM16A suppresses tumor growth and invasion in human lung cancer. PLoS ONE 2015, 10, e0136584. [Google Scholar] [CrossRef] [PubMed]
- House, C.D.; Vaske, C.J.; Schwartz, A.M.; Obias, V.; Frank, B.; Luu, T.; Sarvazyan, N.; Irby, R.; Strausberg, R.L.; Hales, T.G.; et al. Voltage-gated Na+ channel SCN5A is a key regulator of a gene transcriptional network that controls colon cancer invasion. Cancer Res. 2010, 70, 6957–6967. [Google Scholar] [CrossRef] [PubMed]
- Chioni, A.M.; Shao, D.; Grose, R.; Djamgoz, M.B. Protein kinase A and regulation of neonatal Nav1.5 expression in human breast cancer cells: Activity-dependent positive feedback and cellular migration. Int. J. Biochem. Cell Biol. 2010, 42, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.P.; Cabrera, R.M.; Segall, J.E. Tumor cell intravasation. Am. J. Physiol. Cell Physiol. 2016, 311, C1–C14. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.C.; Secrist, H.; Bangur, C.S.; Wang, T.; Zhang, X.; Harlan, D.; Goodman, G.E.; Houghton, R.L.; Persing, D.H.; Zehentner, B.K. Multigene real-time PCR detection of circulating tumor cells in peripheral blood of lung cancer patients. Anticancer Res. 2006, 26, 1567–1575. [Google Scholar] [PubMed]
- Man, Y.; Cao, J.; Jin, S.; Xu, G.; Pan, B.; Shang, L.; Che, D.; Yu, Q.; Yu, Y. Newly identified biomarkers for detecting circulating tumor cells in lung adenocarcinoma. Tohoku J. Exp. Med. 2014, 234, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhi, X.; Zhou, J.; Tao, R.; Zhang, J.; Chen, P.; Røe, O.D.; Sun, L.; Ma, L. Circulating tumor cells as a prognostic and predictive marker in gastrointestinal stromal tumors: A prospective study. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.; Nannmark, U.; Johansson, B.R.; Bagge, U. Lethal deformation of cancer cells in the microcirculation: A potential rate regulator of hematogenous metastasis. Int. J. Cancer 1992, 50, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Lee, A.; Shientag, L.; Yu, J.; Dong, Y.; Kao, G.; Al-Mehdi, A.B.; Bernhard, E.J.; Muschel, R.J. Apoptosis: An early event in metastatic inefficiency. Cancer Res. 2001, 61, 333–338. [Google Scholar] [PubMed]
- Sauter, D.R.; Novak, I.; Pedersen, S.F.; Larsen, E.H.; Hoffmann, E.K. ANO1 (TMEM16A) in pancreatic ductal adenocarcinoma (PDAC). Pflug. Arch. 2015, 467, 1495–1508. [Google Scholar] [CrossRef] [PubMed]
- Furlow, P.W.; Zhang, S.; Soong, T.D.; Halberg, N.; Goodarzi, H.; Mangrum, C.; Wu, Y.G.; Elemento, O.; Tavazoie, S.F. Mechanosensitive pannexin-1 channels mediate microvascular metastatic cell survival. Nat. Cell Biol. 2015, 17, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Martinez, N.; Boire, A.; Deangelis, L.M. Molecular interactions in the development of brain metastases. Int. J. Mol. Sci. 2015, 14, 17157–17167. [Google Scholar] [CrossRef] [PubMed]
- Bittner, S.; Ruck, T.; Schuhmann, M.K.; Herrmann, A.M.; Moha ou Maati, H.; Bobak, N.; Göbel, K.; Langhauser, F.; Stegner, D.; Ehling, P. Endothelial TWIK-related potassium channel-1 (TREK1) regulates immune-cell trafficking into the CNS. Nat. Med. 2013, 19, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Kienast, Y.; von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 2010, 16, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Downie, B.R.; Sanchez, A.; Knotgen, H.; Contreras-Jurado, C.; Gymnopoulos, M.; Weber, C.; Stühmer, W.; Pardo, L.A. Eag1 expression interferes with hypoxia homeostasis and induces angiogenesis in tumors. J. Biol. Chem. 2008, 283, 36234–36240. [Google Scholar] [CrossRef] [PubMed]
- Ouadid-Ahidouch, H.; Ahidouch, A.; Pardo, L.A. Kv10.1 K+ channel: From physiology to cancer. Pflug. Arch. 2016, 468, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, V.; Prasmickaite, L.; Vasiliauskaite, K.; Clancy, T.; Hovig, E. Melanoma brain colonization involves the emergence of a brain-adaptive phenotype. Oncoscience 2014, 1, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Neman, J.; Termini, J.; Wilczynski, S.; Vaidehi, N.; Choy, C.; Kowolik, C.M.; Li, H.; Hambrecht, A.C.; Roberts, E.; Jandial, R. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc. Natl. Acad. Sci. USA 2014, 111, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Black, K.L.; Ningaraj, N.S. Modulation of brain tumor capillaries for enhanced drug delivery selectively to brain tumor. Cancer Control 2004, 11, 165–173. [Google Scholar] [PubMed]
- Ningaraj, N.S.; Rao, M.; Black, K.L. Calcium-dependent potassium channels as a target protein for modulation of the blood-brain tumor barrier. Drug News Perspect. 2003, 16, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Yuan, X.; Ko, M.K.; Yin, D.; Sacapano, M.R.; Wang, X.; Konda, B.M.; Espinoza, A.; Prosolovich, K.; Ong, J.M.; et al. Calcium-activated potassium channels mediated blood-brain tumor barrier opening in a rat metastatic brain tumor model. Mol. Cancer 2007, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gu, Y.T.; Xue, Y.X. Bradykinin-induced blood-brain tumor barrier permeability increase is mediated by adenosine 5′-triphosphate-sensitive potassium channel. Brain Res. 2007, 1144, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Golanov, A.V.; Banov, S.M.; Il’yalov, S.R.; Trunin, Y.Y.; Maryashev, S.A.; Vetlova, E.R.; Osinov, I.K.; Kostyuchenko, V.V.; Dalechina, A.V.; Durgaryan, A.A. Overall survival and intracranial relapse in patients with brain metastases after γ knife radiosurgery alone. Zhurnal Voprosy Neirokhirurgii Imeni NN Burdenko 2016, 80, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Eckert, F.; Gani, C.; Bamberg, M.; Muller, A.C. Cerebral metastases in extrapulmonary cell carcinoma. Implications for the use of prophylactic cranial irradiation. Strahlenther. Onkol. 2012, 188, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Heise, N.; Palme, D.; Misovic, M.; Koka, S.; Rudner, J.; Lang, F.; Salih, H.R.; Huber, S.M.; Henke, G. Non-selective cation channel-mediated Ca2+-entry and activation of Ca2+/calmodulin-dependent kinase II contribute to G2/M cell cycle arrest and survival of irradiated leukemia cells. Cell. Physiol. Biochem. 2010, 26, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, D.; Misovic, M.; Szteyn, K.; Shumilina, E.; Rudner, J.; Huber, S.M. Targeting TRPM2 channels impairs radiation-induced cell cycle arrest and fosters cell death of T cell leukemia cells in a Bcl-2-dependent manner. Oxid. Med. Cell. Longev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Palme, D.; Misovic, M.; Schmid, E.; Klumpp, D.; Salih, H.R.; Rudner, J.; Huber, S.M. Kv3.4 potassium channel-mediated electrosignaling controls cell cycle and survival of irradiated leukemia cells. Pflug. Arch. 2013, 465, 1209–1221. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.M.; Misovic, M.; Mayer, C.; Rodemann, H.P.; Dittmann, K. EGFR-mediated stimulation of sodium/glucose cotransport promotes survival of irradiated human A549 lung adenocarcinoma cells. Radiother. Oncol. 2012, 103, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Stegen, B.; Butz, L.; Klumpp, L.; Zips, D.; Dittmann, K.; Ruth, P.; Huber, S.M. Ca2+-activated IK K+ channel blockade radiosensitizes glioblastoma cells. Mol. Cancer Res. 2015, 13, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Gibhardt, C.S.; Roth, B.; Schroeder, I.; Fuck, S.; Becker, P.; Jakob, B.; Fournier, C.; Moroni, A.; Thiel, G. X-ray irradiation activates K+ channels via H2O2 signaling. Sci. Rep. 2015, 5, 13861. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.; Gibhardt, C.S.; Becker, P.; Gebhardt, M.; Knoop, J.; Fournier, C.; Moroni, A.; Thiel, G. Low-dose photon irradiation alters cell differentiation via activation of hIK channels. Pflug. Arch. 2015, 467, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Stegen, B.; Klumpp, L.; Misovic, M.; Edalat, L.; Eckert, M.; Klumpp, D.; Ruth, P.; Huber, S.M. K+ channels signaling in irradiated tumor cells. Eur. Biophys. J. 2016. [Google Scholar] [CrossRef] [PubMed]
- Steinle, M.; Palme, D.; Misovic, M.; Rudner, J.; Dittmann, K.; Lukowski, R.; Ruth, P.; Huber, S.M. Ionizing radiation induces migration of glioblastoma cells by activating BK K+ channels. Radiother. Oncol. 2011, 101, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Edalat, L.; Stegen, B.; Klumpp, L.; Haehl, E.; Schilbach, K.; Lukowski, R.; Kühnle, M.; Bernhardt, G.; Buschauer, A.; Zips, D.; et al. BK K+ channel blockade inhibits radiation-induced migration/brain infiltration of glioblastoma cells. Oncotarget 2016, 7, 14259–14278. [Google Scholar] [PubMed]
- Braun, N.; Klumpp, D.; Hennenlotter, J.; Bedke, J.; Duranton, C.; Bleif, M.; Huber, S.M. UCP-3 uncoupling protein confers hypoxia resistance to renal epithelial cells and is upregulated in renal cell carcinoma. Sci. Rep. 2015, 5, 13450. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, B.; Tang, S.; Guo, H.; Li, W.; Huang, X.; Yan, W.; Zou, F. Mitochondrial KATPchannels control glioma radioresistance by regulating ROS-induced ERK activation. Mol. Neurobiol. 2015, 52, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.J. CXCR4 and glioblastoma. Anticancer Agents Med. Chem. 2015, 16, 59–74. [Google Scholar] [CrossRef]
- Sontheimer, H. An unexpected role for ion channels in brain tumor metastasis. Exp. Biol. Med. 2008, 233, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Sin, W.C.; Harris, A.L.; Naus, C.C. Gap junctions modulate glioma invasion by direct transfer of microRNA. Oncotarget 2015, 6, 15566–15577. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; McKenna, F.; Rowe, I.C.; Ashford, M.L. The effects of neuroleptic and tricyclic compounds on BKCa channel activity in rat isolated cortical neurones. Br. J. Pharmacol. 1997, 121, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Korpi, E.R.; Kleinman, J.E.; Costakos, D.T.; Linnoila, M.; Wyatt, R.J. Reduced haloperidol in the post-mortem brains of haloperidol-treated patients. Psychiatry Res. 1984, 11, 259–269. [Google Scholar] [CrossRef]
- Huang, C.L.; Ruskin, B.H. Determination of serum chlorpromazine metabolites in psychotic patients. J. Nerv. Ment. Dis. 1964, 139, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Maezawa, I.; Jenkins, D.P.; Jin, B.E.; Wulff, H. Microglial KCa3.1 channels as a potential therapeutic target for Alzheimer’s disease. Int. J. Alzheimers Dis. 2012, 2012, 868972. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Ufodiama, C.; Watt, I.; Bland, M.; Brackenbury, W.J. Therapeutic value of voltage-gated sodium channel inhibitors in breast, colorectal, and prostate cancer: A Systematic. Rev. Front. Pharmacol. 2015, 6, 273. [Google Scholar] [CrossRef] [PubMed]
- Fairhurst, C.; Watt, I.; Martin, F.; Bland, M.; Brackenbury, W.J. Sodium channel-inhibiting drugs and survival of breast, colon and prostate cancer: A population-based study. Sci. Rep. 2015, 5, 16758. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klumpp, L.; Sezgin, E.C.; Eckert, F.; Huber, S.M. Ion Channels in Brain Metastasis. Int. J. Mol. Sci. 2016, 17, 1513. https://doi.org/10.3390/ijms17091513
Klumpp L, Sezgin EC, Eckert F, Huber SM. Ion Channels in Brain Metastasis. International Journal of Molecular Sciences. 2016; 17(9):1513. https://doi.org/10.3390/ijms17091513
Chicago/Turabian StyleKlumpp, Lukas, Efe C. Sezgin, Franziska Eckert, and Stephan M. Huber. 2016. "Ion Channels in Brain Metastasis" International Journal of Molecular Sciences 17, no. 9: 1513. https://doi.org/10.3390/ijms17091513
APA StyleKlumpp, L., Sezgin, E. C., Eckert, F., & Huber, S. M. (2016). Ion Channels in Brain Metastasis. International Journal of Molecular Sciences, 17(9), 1513. https://doi.org/10.3390/ijms17091513