
Monitoring the Response of Hyperbilirubinemia in the Mouse Brain by In Vivo Bioluminescence Imaging


 ,
,  ,
,
Abstract
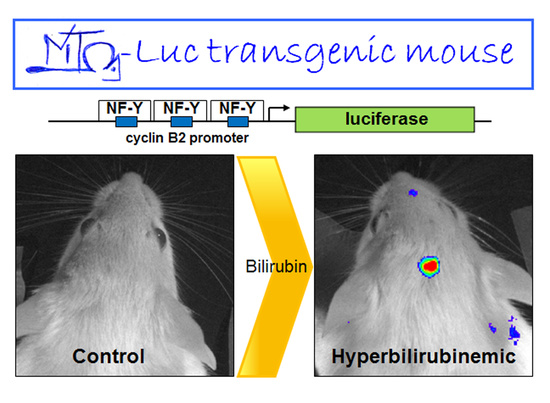
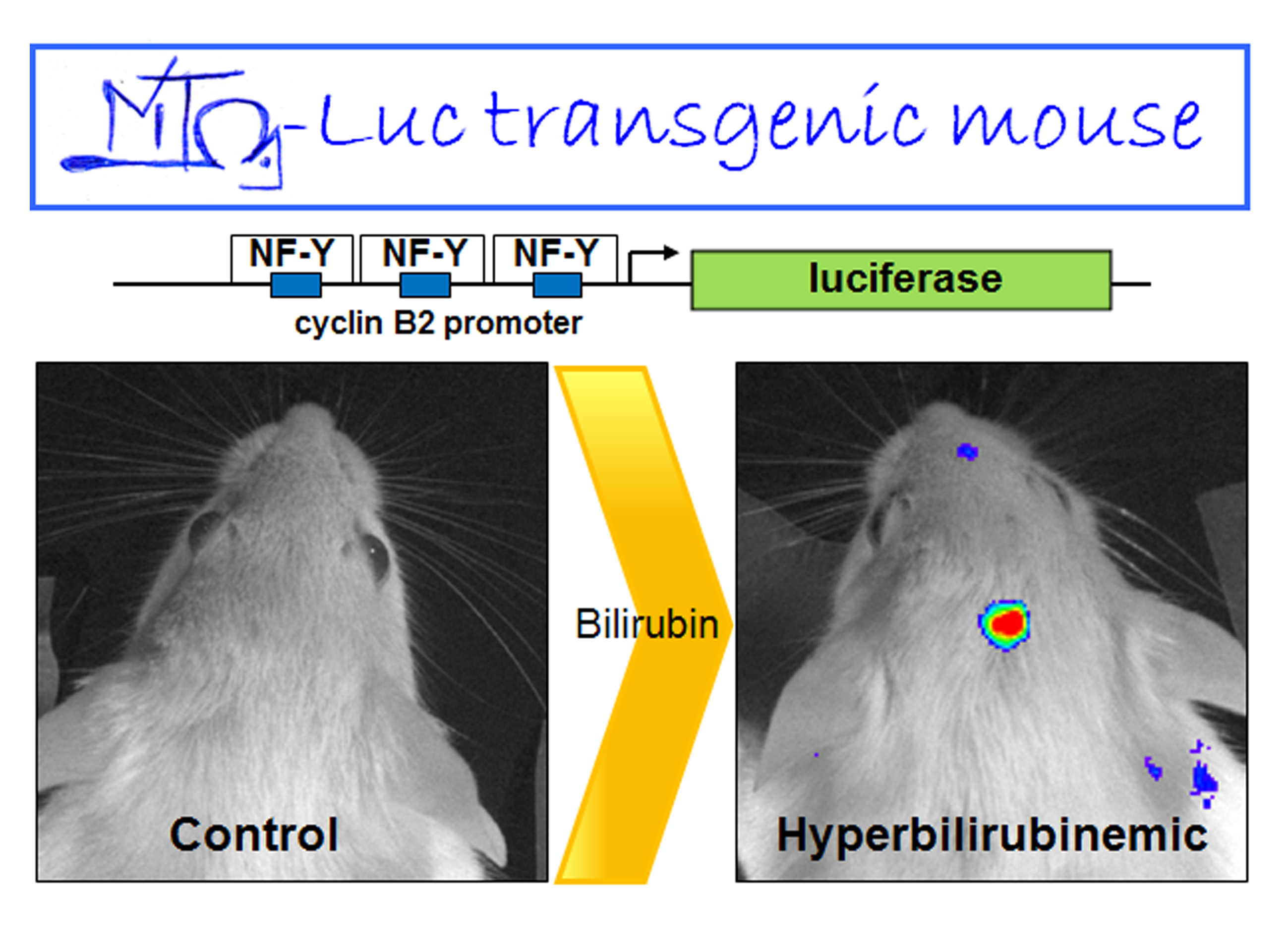
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
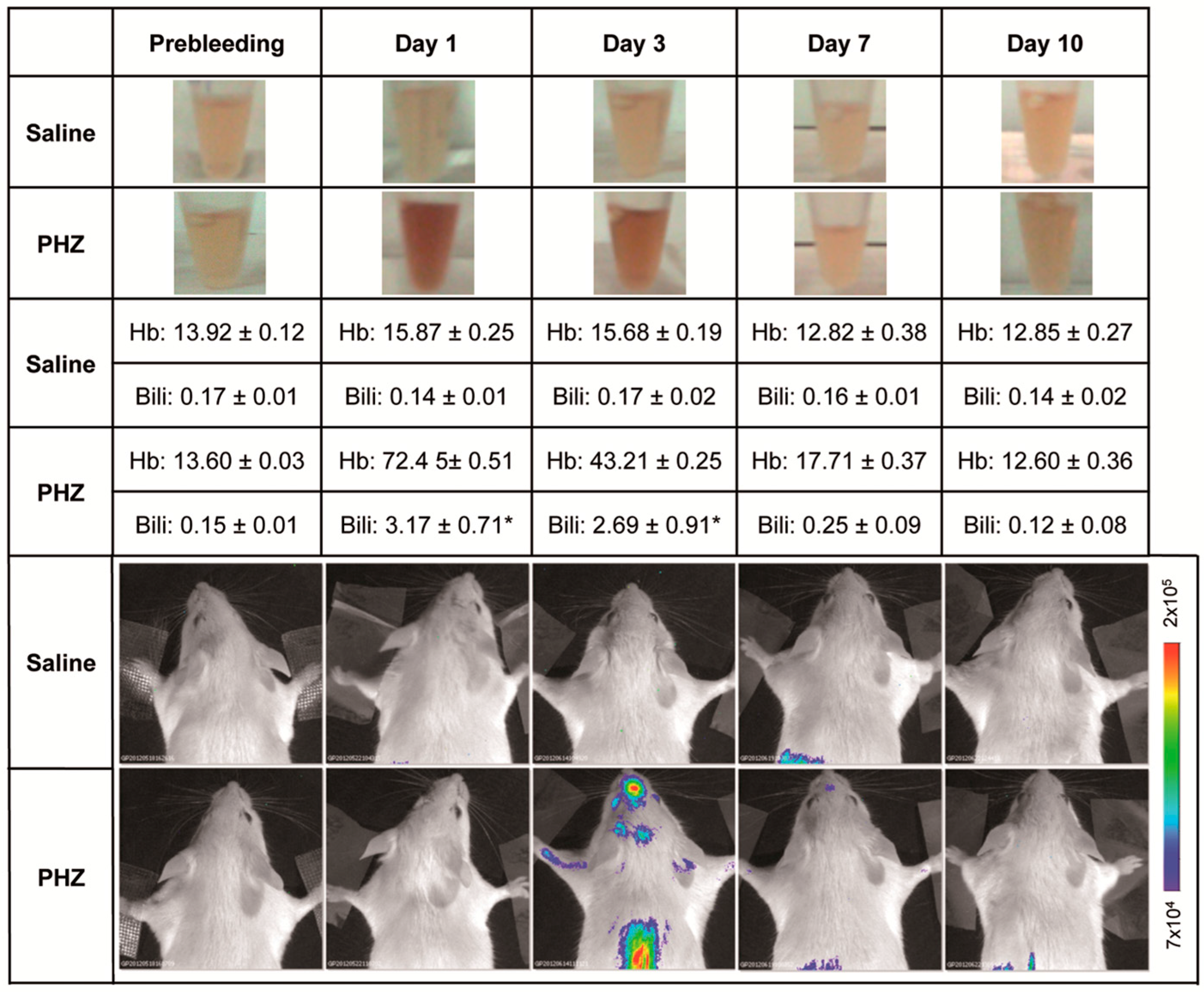
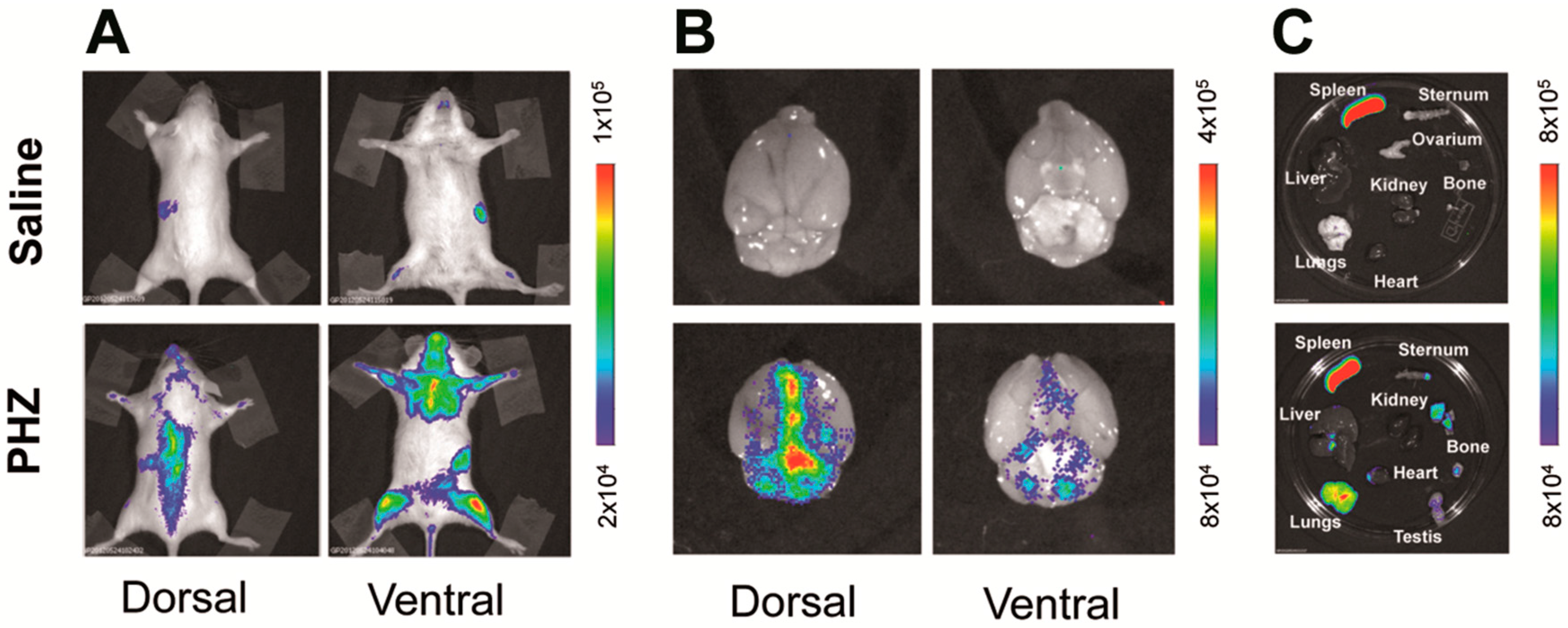
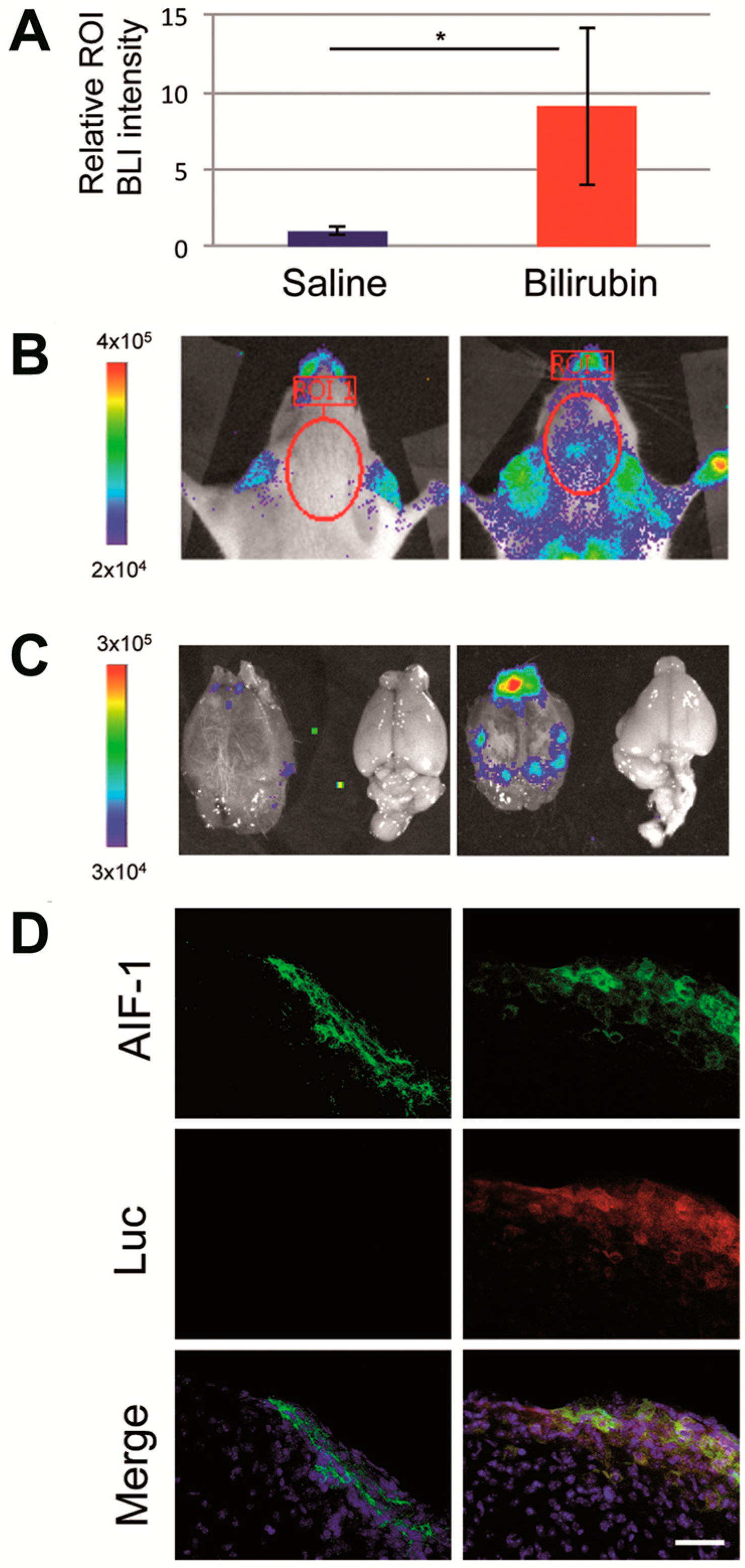
2.1. Hyperbilirubinemia Induces Bioluminescence in the Brain of MITO-Luc Mice
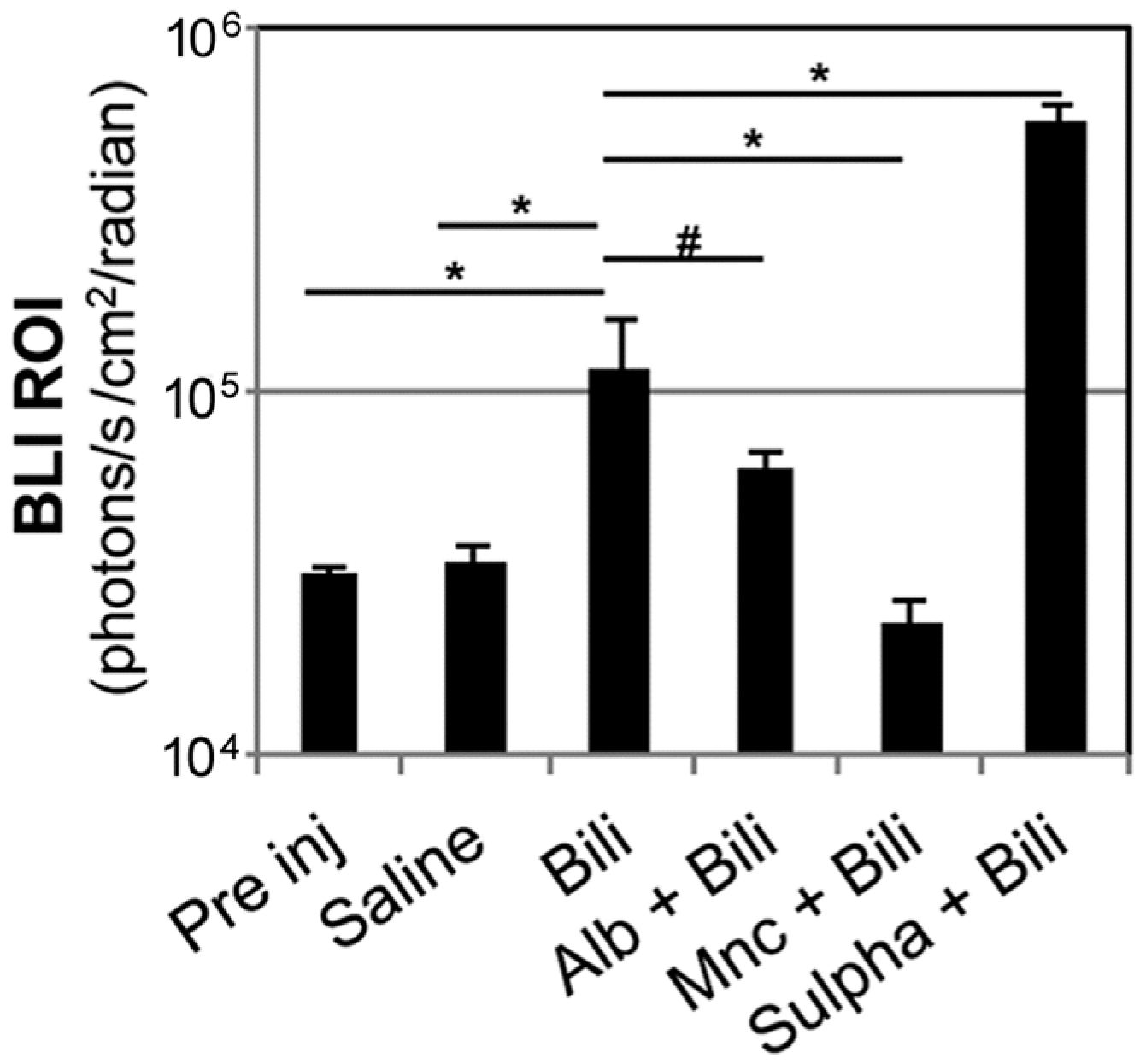
2.2. Modulation of Bilirubin/Albumin Binding Impinges on Bilirubin-Induced Bioluminescence in MITO-Luc Mice
2.3. Minocycline Treatment Blunts Hyperbilirubinemia-Induced Bioluminescence in MITO-Luc Mice
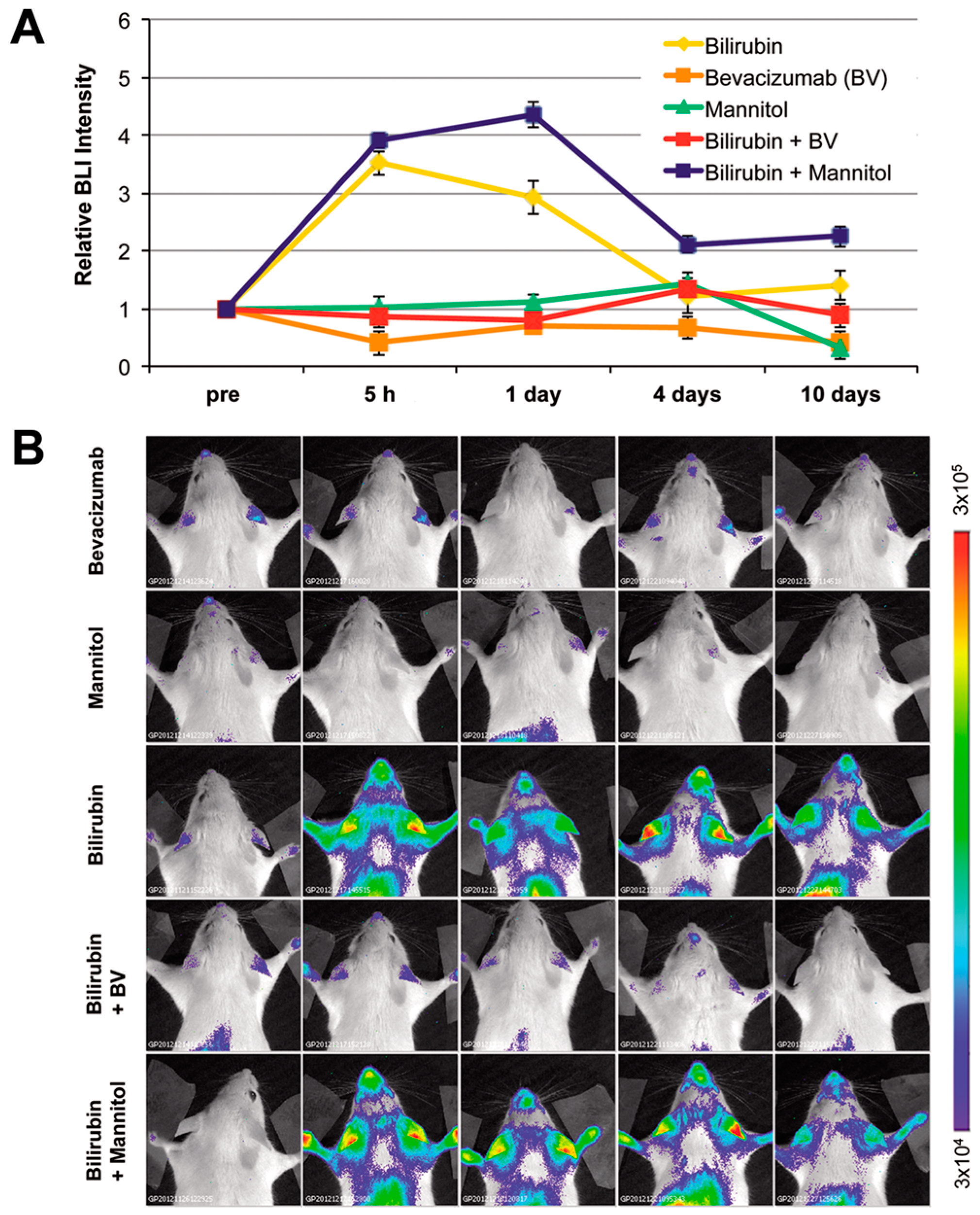
2.4. Modulation of Blood–Brain Barrier Permeability Affects Bioluminescence in the Brain Region in Hyperbilirubinemic MITO-Luc Mice
3. Discussion
4. Materials and Methods
4.1. Experimental Animal Procedures
4.2. In Vivo and Ex Vivo Optical Bioluminescence Imaging
4.3. Immunohistological Analysis
4.4. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BIND | Bilirubin-induced neurological dysfunction |
| NF-Y | Nuclear factor-Y |
| Alb | Albumin |
| BBB | Blood–brain barrier |
| BLI | Bioluminescence imaging |
| Mnc | Minocycline |
| PHZ | Phenylhydrazine |
| UCB | Unconjugated bilirubin |
References
- Shapiro, S.M. Definition of the clinical spectrum of kernicterus and bilirubin-induced neurologic dysfunction (BIND). J. Perinatol. 2005, 25, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, V.K.; Johnson-Hamerman, L. The clinical syndrome of bilirubin-induced neurologic dysfunction. Semin. Fetal Neonatal Med. 2015, 20, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Gazzin, S.; Strazielle, N.; Tiribelli, C.; Ghersi-Egea, J.F. Transport and metabolism at blood–brain interfaces and in neural cells: Relevance to bilirubin-induced encephalopathy. Front. Pharmacol. 2012, 3, 89. [Google Scholar] [CrossRef] [PubMed]
- Palmela, I.; Sasaki, H.; Cardoso, F.L.; Moutinho, M.; Kim, K.S.; Brites, D.; Brito, M.A. Time-dependent dual effects of high levels of unconjugated bilirubin on the human blood–brain barrier lining. Front. Cell. Neurosci. 2012, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Wennberg, R.P. The blood–brain barrier and bilirubin encephalopathy. Cell. Mol. Neurobiol. 2000, 20, 97–109. [Google Scholar] [PubMed]
- Ostrow, J.D.; Pascolo, L.; Brites, D.; Tiribelli, C. Molecular basis of bilirubin-induced neurotoxicity. Trends Mol. Med. 2004, 10, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Genc, S.; Genc, K.; Kumral, A.; Baskin, H.; Ozkan, H. Bilirubin is cytotoxic to rat oligodendrocytes in vitro. Brain Res. 2003, 985, 135–141. [Google Scholar] [PubMed]
- Silva, R.F.; Rodrigues, C.M.; Brites, D. Rat cultured neuronal and glial cells respond differently to toxicity of unconjugated bilirubin. Pediatr. Res. 2002, 51, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, C.; Capone, C.; Ranieri, S.C.; Fusco, S.; Calabrese, V.; Eboli, M.L.; Preziosi, P.; Galeotti, T.; Pani, G. Bilirubin as an endogenous modulator of neurotrophin redox signaling. J. Neurosci. Res. 2008, 86, 2235–2249. [Google Scholar] [PubMed]
- Deganuto, M.; Cesaratto, L.; Bellarosa, C.; Calligaris, R.; Vilotti, S.; Renzone, G.; Foti, R.; Scaloni, A.; Gustincich, S.; Quadrifoglio, F.; et al. A proteomic approach to the bilirubin-induced toxicity in neuronal cells reveals a protective function of DJ-1 protein. Proteomics 2010, 10, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.; Falcão, A.S.; Silva, R.F.; Gordo, A.C.; Gama, M.J.; Brito, M.A.; Brites, D. Inflammatory signalling pathways involved in astroglial activation by unconjugated bilirubin. J. Neurochem. 2006, 96, 1667–1679. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.L.; Vaz, A.R.; Barateiro, A.; Falcão, A.S.; Fernandes, A.; Brito, M.A.; Silva, R.F.; Brites, D. Features of bilirubin-induced reactive microglia: From phagocytosis to inflammation. Neurobiol. Dis. 2010, 40, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.C.; Shapiro, S.M. A new animal model of hemolytic hyperbilirubinemia-induced bilirubin encephalopathy (kernicterus). Pediatr. Res. 2008, 64, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Mejia, G.B.; Sanz, C.R.; Avila, M.M.; Peraza, A.V.; Guzmán, D.C.; Olguín, H.J.; Ramírez, A.M.; Cruz, E.G. Experimental hemolysis model to study bilirubin encephalopathy in rat brain. J. Neurosci. Methods 2008, 168, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Amini, N.; Vousooghi, N.; Soleimani, M.; Samadikuchaksaraei, A.; Akbari, M.; Safakheil, H.; Atafimanesh, P.; Shahbazi, A.; Brouki Milan, P.; Ramezani, S.; et al. A new rat model of neonatal bilirubin encephalopathy (kernicterus). J. Pharmacol. Toxicol. Methods 2016, 84, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Goeman, F.; Manni, I.; Artuso, S.; Ramachandran, B.; Toietta, G.; Bossi, G.; Rando, G.; Cencioni, C.; Germoni, S.; Straino, S.; et al. Molecular imaging of nuclear factor-Y transcriptional activity maps proliferation sites in live animals. Mol. Biol. Cell 2012, 23, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, N.; Manni, I.; Vantaggiato, C.; Delledonne, G.A.; Gentileschi, M.P.; Maggi, A.; Piaggio, G.; Ciana, P. In vivo imaging of cell proliferation for a dynamic, whole body, analysis of undesired drug effects. Toxicol. Sci. 2015, 145, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Courties, G.; Herisson, F.; Sager, H.B.; Heidt, T.; Ye, Y.; Wei, Y.; Sun, Y.; Severe, N.; Dutta, P.; Scharff, J.; et al. Ischemic stroke activates hematopoietic bone marrow stem cells. Circ. Res. 2015, 116, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.; Yadav, N.; Singh, P.; Bansode, F.; Singh, R. Phenylhydrazine induced toxicity: A review on its haematotoxicity. Int. J. Basic Appl. Med. Sci. 2012, 2, 86–91. [Google Scholar]
- Berger, J. Phenylhydrazine haematotoxicity. J. Appl. Biomed. 2007, 5, 125–130. [Google Scholar]
- Naughton, B.A.; Dornfest, B.S.; Bush, M.E.; Carlson, C.A.; Lapin, D.M. Immune activation is associated with phenylhydrazine-induced anemia in the rat. J. Lab. Clin. Med. 1990, 116, 498–507. [Google Scholar] [PubMed]
- Sato, H.; Sakairi, T.; Fujimura, H.; Sugimoto, J.; Kume, E.; Kitamura, K.; Takahashi, K. Hematological and morphological investigation of thrombogenic mechanisms in the lungs of phenylhydrazine-treated rats. Exp. Toxicol. Pathol. 2013, 65, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Liaury, K.; Miyaoka, T.; Tsumori, T.; Furuya, M.; Wake, R.; Ieda, M.; Tsuchie, K.; Taki, M.; Ishihara, K.; Tanra, A.J.; et al. Morphological features of microglial cells in the hippocampal dentate gyrus of Gunn rat: A possible schizophrenia animal model. J. Neuroinflamm. 2012, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Ladeby, R.; Wirenfeldt, M.; Garcia-Ovejero, D.; Fenger, C.; Dissing-Olesen, L.; Dalmau, I.; Finsen, B. Microglial cell population dynamics in the injured adult central nervous system. Brain Res. Rev. 2005, 48, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Brites, D. The evolving landscape of neurotoxicity by unconjugated bilirubin: Role of glial cells and inflammation. Front. Pharmacol. 2012, 3, 88. [Google Scholar] [CrossRef] [PubMed]
- Hosono, S.; Ohno, T.; Kimoto, H.; Nagoshi, R.; Shimizu, M.; Nozawa, M. Effects of albumin infusion therapy on total and unbound bilirubin values in term infants with intensive phototherapy. Pediatr. Int. 2001, 43, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Ahlfors, C.E. Pre exchange transfusion administration of albumin: An overlooked adjunct in the treatment of severe neonatal jaundice? Indian Pediatr. 2010, 47, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Geiger, A.S.; Rice, A.C.; Shapiro, S.M. Minocycline blocks acute bilirubin-induced neurological dysfunction in jaundiced Gunn rats. Neonatology 2007, 92, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.C.; Chiou, V.L.; Zuckoff, S.B.; Shapiro, S.M. Profile of minocycline neuroprotection in bilirubin-induced auditory system dysfunction. Brain Res. 2011, 1368, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Mesa, N.; Zarzuelo, A.; Gálvez, J. Minocycline: Far beyond an antibiotic. Br. J. Pharmacol. 2013, 169, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Plane, J.M.; Shen, Y.; Pleasure, D.E.; Deng, W. Prospects for minocycline neuroprotection. Arch. Neurol. 2010, 67, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Yenari, M.A.; Xu, L.; Tang, X.N.; Qiao, Y.; Giffard, R.G. Microglia potentiate damage to blood–brain barrier constituents: Improvement by minocycline in vivo and in vitro. Stroke 2006, 37, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Daood, M.J.; Hoyson, M.; Watchko, J.F. Lipid peroxidation is not the primary mechanism of bilirubin-induced neurologic dysfunction in jaundiced Gunn rat pups. Pediatr. Res. 2012, 72, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Gumerlock, M.K.; Belshe, B.D.; Madsen, R.; Watts, C. Osmotic blood–brain barrier disruption and chemotherapy in the treatment of high grade malignant glioma: Patient series and literature review. J. Neurooncol. 1992, 12, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Gonzales-Portillo, G.S.; Sanberg, P.R.; Franzblau, M.; Gonzales-Portillo, C.; Diamandis, T.; Staples, M.; Sanberg, C.D.; Borlongan, C.V. Mannitol-enhanced delivery of stem cells and their growth factors across the blood–brain barrier. Cell Transplant. 2014, 23, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Boockvar, J.A.; Tsiouris, A.J.; Hofstetter, C.P.; Kovanlikaya, I.; Fralin, S.; Kesavabhotla, K.; Seedial, S.M.; Pannullo, S.C.; Schwartz, T.H.; Stieg, P.; et al. Safety and maximum tolerated dose of superselective intraarterial cerebral infusion of bevacizumab after osmotic blood–brain barrier disruption for recurrent malignant glioma. Clinical article. J. Neurosurg. 2011, 114, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Riordan, S.M.; Bittel, D.C.; Le Pichon, J.B.; Gazzin, S.; Tiribelli, C.; Watchko, J.F.; Wennberg, R.P.; Shapiro, S.M. A hypothesis for using pathway genetic load analysis for understanding complex outcomes in bilirubin encephalopathy. Front. Neurosci. 2016, 10, 376. [Google Scholar] [CrossRef] [PubMed]
- Stolp, H.B.; Ek, C.J.; Johansson, P.A.; Dziegielewska, K.M.; Potter, A.M.; Habgood, M.D.; Saunders, N.R. Effect of minocycline on inflammation-induced damage to the blood–brain barrier and white matter during development. Eur. J. Neurosci. 2007, 26, 3465–3474. [Google Scholar] [CrossRef] [PubMed]
- Gordo, A.C.; Falcão, A.S.; Fernandes, A.; Brito, M.A.; Silva, R.F.; Brites, D. Unconjugated bilirubin activates and damages microglia. J. Neurosci. Res. 2006, 84, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Brites, D. Bilirubin injury to neurons and glial cells: New players, novel targets, and newer insights. Semin. Perinatol. 2011, 35, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Watchko, J.F. Kernicterus and the molecular mechanisms of bilirubin-induced CNS injury in newborns. Neuromol. Med. 2006, 8, 513–529. [Google Scholar] [CrossRef]
- Deng, S.X.; Panahian, N.; James, H.; Gelbard, H.A.; Federoff, H.J.; Dewhurst, S.; Epstein, L.G. Luciferase: A sensitive and quantitative probe for blood–brain barrier disruption. J. Neurosci. Methods 1998, 83, 159–164. [Google Scholar] [CrossRef]
- Alayoff, A.; Kapitulnik, J.; Konijn, A.; Kaufmann, N.A.; Blondheim, S.H. Bilirubin binding capacity of albumin isolated from cord-blood serum is less than that from serum of adults. Clin. Chem. 1980, 26, 738–740. [Google Scholar] [PubMed]
- Valledor, A.F.; Borràs, F.E.; Cullell-Young, M.; Celada, A. Transcription factors that regulate monocyte/macrophage differentiation. J. Leukoc. Biol. 1998, 63, 405–417. [Google Scholar] [PubMed]
- Gomez-Nicola, D.; Perry, V.H. Microglial dynamics and role in the healthy and diseased brain: A paradigm of functional plasticity. Neuroscientist 2015, 21, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Lalancette-Hébert, M.; Gowing, G.; Simard, A.; Weng, Y.C.; Kriz, J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci. 2007, 27, 2596–2605. [Google Scholar] [CrossRef] [PubMed]
- Olmos-Alonso, A.; Schetters, S.T.; Sri, S.; Askew, K.; Mancuso, R.; Vargas-Caballero, M.; Holscher, C.; Perry, V.H.; Gomez-Nicola, D. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain 2016, 139, 891–907. [Google Scholar] [CrossRef] [PubMed]
- Vidiri, A.; Pace, A.; Fabi, A.; Maschio, M.; Latagliata, G.M.; Anelli, V.; Piludu, F.; Carapella, C.M.; Giovinazzo, G.; Marzi, S. Early perfusion changes in patients with recurrent high-grade brain tumor treated with bevacizumab: Preliminary results by a quantitative evaluation. J. Exp. Clin. Cancer Res. 2012, 31, 33. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Nemeño, J.G.; Lee, J.I. Repositioning bevacizumab: A promising therapeutic strategy for cartilage regeneration. Tissue Eng. Part B Rev. 2016, 22, 341–357. [Google Scholar] [CrossRef] [PubMed]
- De Pasquale, M.D.; Castellano, A.; de Sio, L.; de Laurentis, C.; Mastronuzzi, A.; Serra, A.; Cozza, R.; Jenkner, A.; de Ioris, M.A. Bevacizumab in pediatric patients: How safe is it? Anticancer Res. 2011, 31, 3953–3957. [Google Scholar] [PubMed]
- Ghodsi, A.; Stein, C.; Derksen, T.; Martins, I.; Anderson, R.D.; Davidson, B.L. Systemic hyperosmolality improves β-glucuronidase distribution and pathology in murine MPS VII brain following intraventricular gene transfer. Exp. Neurol. 1999, 160, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Baldari, S.; di Rocco, G.; Trivisonno, A.; Samengo, D.; Pani, G.; Toietta, G. Promotion of survival and engraftment of transplanted adipose tissue-derived stromal and vascular cells by overexpression of manganese superoxide dismutase. Int. J. Mol. Sci. 2016, 17, 1082. [Google Scholar] [CrossRef] [PubMed]
- Di Rocco, G.; Gentile, A.; Antonini, A.; Truffa, S.; Piaggio, G.; Capogrossi, M.C.; Toietta, G. Analysis of biodistribution and engraftment into the liver of genetically modified mesenchymal stromal cells derived from adipose tissue. Cell Transplant. 2012, 21, 1997–2008. [Google Scholar] [CrossRef] [PubMed]
- Leone, L.; Fusco, S.; Mastrodonato, A.; Piacentini, R.; Barbati, S.A.; Zaffina, S.; Pani, G.; Podda, M.V.; Grassi, C. Epigenetic modulation of adult hippocampal neurogenesis by extremely low-frequency electromagnetic fields. Mol. Neurobiol. 2014, 49, 1472–1486. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manni, I.; Di Rocco, G.; Fusco, S.; Leone, L.; Barbati, S.A.; Carapella, C.M.; Grassi, C.; Piaggio, G.; Toietta, G. Monitoring the Response of Hyperbilirubinemia in the Mouse Brain by In Vivo Bioluminescence Imaging. Int. J. Mol. Sci. 2017, 18, 50. https://doi.org/10.3390/ijms18010050
Manni I, Di Rocco G, Fusco S, Leone L, Barbati SA, Carapella CM, Grassi C, Piaggio G, Toietta G. Monitoring the Response of Hyperbilirubinemia in the Mouse Brain by In Vivo Bioluminescence Imaging. International Journal of Molecular Sciences. 2017; 18(1):50. https://doi.org/10.3390/ijms18010050
Chicago/Turabian StyleManni, Isabella, Giuliana Di Rocco, Salvatore Fusco, Lucia Leone, Saviana Antonella Barbati, Carmine Maria Carapella, Claudio Grassi, Giulia Piaggio, and Gabriele Toietta. 2017. "Monitoring the Response of Hyperbilirubinemia in the Mouse Brain by In Vivo Bioluminescence Imaging" International Journal of Molecular Sciences 18, no. 1: 50. https://doi.org/10.3390/ijms18010050
APA StyleManni, I., Di Rocco, G., Fusco, S., Leone, L., Barbati, S. A., Carapella, C. M., Grassi, C., Piaggio, G., & Toietta, G. (2017). Monitoring the Response of Hyperbilirubinemia in the Mouse Brain by In Vivo Bioluminescence Imaging. International Journal of Molecular Sciences, 18(1), 50. https://doi.org/10.3390/ijms18010050