
Targeting IRES-Mediated p53 Synthesis for Cancer Diagnosis and Therapeutics

Abstract
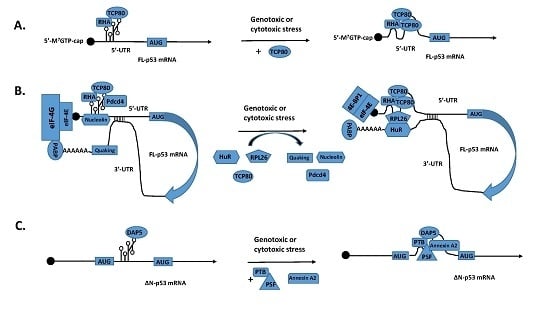
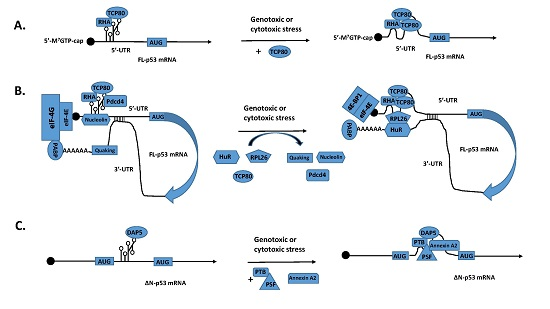
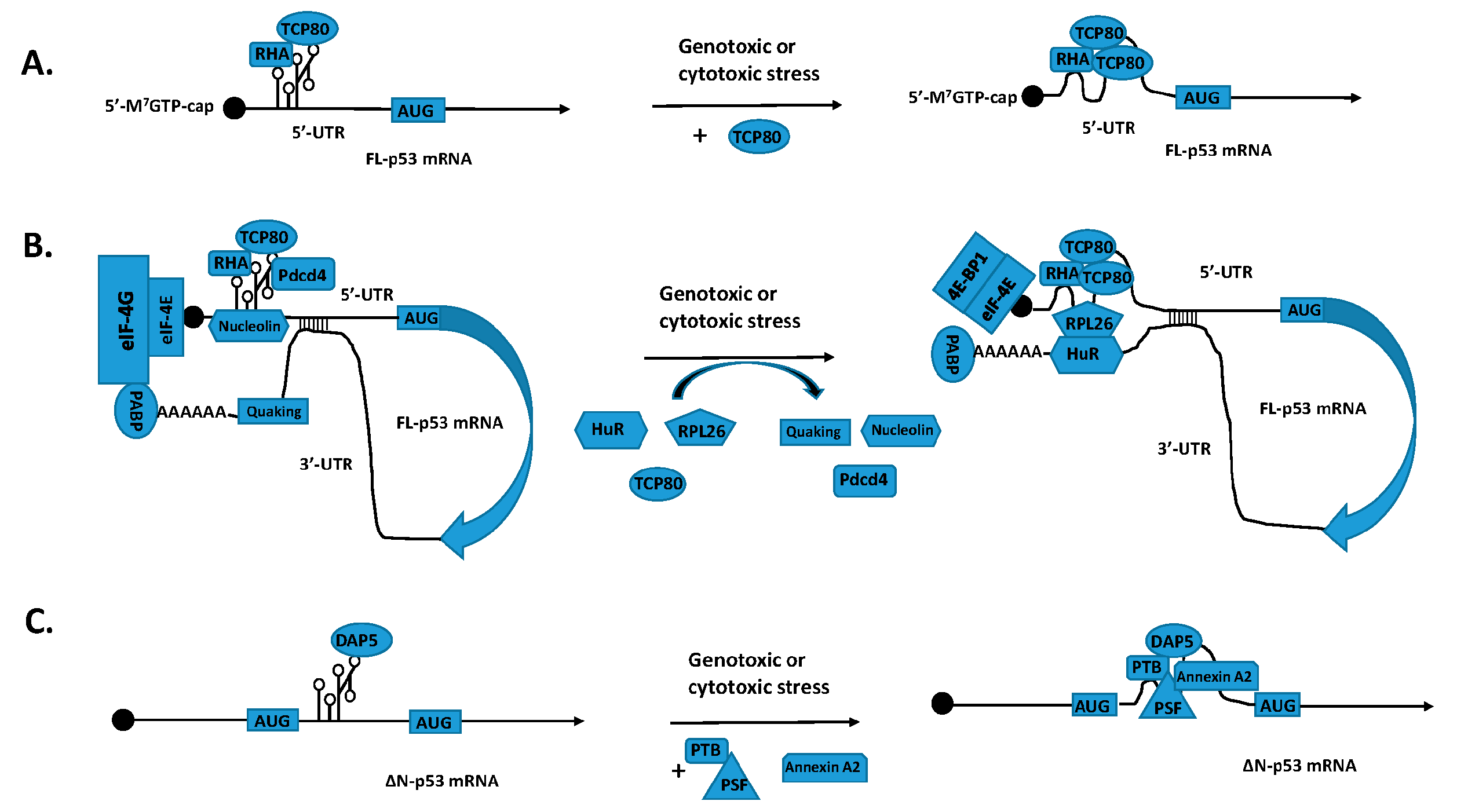
:
{kind=link}
{kind=link}
1. Introduction
2. A Switch from Cap-Dependent Translation to IRES-Mediated Translation of p53 mRNA Following DNA Damage and Discovery of the p53 IRES
3. Identification of p53 ITAFs That Regulate IRES-Mediated Translation of p53 mRNA
4. Regulation of p53 mRNA Translation by its 3′-UTR through the Interaction between its 5′- and 3′-UTR binding proteins
5. Altered Expression of p53 ITAFs in Cancer Cells with Defective Induction of p53 and Its Implication in Tumorigenesis and Cancer Diagnosis
6. Targeting IRES-Mediated p53 Synthesis for Cancer Therapeutics
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Giaccia, A.J.; Kastan, M.B. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes Dev. 1998, 12, 2973–2983. [Google Scholar] [CrossRef]
- Halaby, M.J.; Yang, D.Q. p53 translational control: A new facet of p53 regulation and its implication for tumorigenesis and cancer therapeutics. Gene 2007, 395, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Grover, R.; Candeias, M.M.; Fahraeus, R.; Das, S. p53 and little brother p53/47: Linking IRES activities with protein functions. Oncogene 2009, 28, 2766–2772. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.Q.; Halaby, M.J.; Zhang, Y. The identification of an internal ribosomal entry site in the 5′-untranslated region of p53 mRNA provides a novel mechanism for the regulation of its translation following DNA damage. Oncogene 2006, 25, 4613–4619. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.S.; Grover, R.; Das, S. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep. 2006, 7, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Halaby, M.J.; Harris, B.R.; Miskimins, W.K.; Cleary, M.P.; Yang, D.Q. Deregulation of IRES-mediated p53 translation in cancer cells with defective p53 response to DNA damage. Mol. Cell Biol. 2015, 35, 4006–4017. [Google Scholar] [CrossRef] [PubMed]
- Halaby, M.J.; Li, Y.; Harris, B.R.; Jiang, S.; Miskimins, W.K.; Cleary, M.P.; Yang, D.Q. Translational Control Protein 80 Stimulates IRES-Mediated Translation of p53 mRNA in Response to DNA Damage. BioMed Res. Int. 2015, 2015, 708158. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987, 15, 8125–8148. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Raught, B.; Sonenberg, N. eIF4 initiation factors: Effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 1999, 68, 913–963. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Brunn, G.J.; Lawrence, J.C., Jr. Mutational analysis of sites in the translational regulator, PHAS-I, that are selectively phosphorylated by mTOR. FEBS Lett. 1999, 453, 387–390. [Google Scholar] [CrossRef]
- Yang, D.Q.; Kastan, M.B. Participation of ATM in insulin signalling through phosphorylation of eIF-4E-binding protein 1. Nat. Cell Biol. 2000, 2, 893–898. [Google Scholar] [PubMed]
- Holcik, M.; Sonenberg, N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991, 51 Pt 1, 6304–6311. [Google Scholar] [CrossRef] [PubMed]
- Mazan-Mamczarz, K.; Galban, S.; Lopez de Silanes, I.; Martindale, J.L.; Atasoy, U.; Keene, J.D.; Gorospe, M. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc. Natl. Acad. Sci. USA 2003, 100, 8354–8359. [Google Scholar] [CrossRef] [PubMed]
- Mosner, J.; Mummenbrauer, T.; Bauer, C.; Sczakiel, G.; Grosse, F.; Deppert, W. Negative feedback regulation of wild-type p53 biosynthesis. Embo J. 1995, 14, 4442–4449. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Benchimol, S. Participation of the human p53 3′UTR in translational repression and activation following gamma-irradiation. Embo J. 1997, 16, 4117–4125. [Google Scholar] [CrossRef] [PubMed]
- Candeias, M.M.; Powell, D.J.; Roubalova, E.; Apcher, S.; Bourougaa, K.; Vojtesek, B.; Bruzzoni-Giovanelli, H.; Fahraeus, R. Expression of p53 and p53/47 are controlled by alternative mechanisms of messenger RNA translation initiation. Oncogene 2006, 25, 6936–6947. [Google Scholar] [CrossRef] [PubMed]
- Sayan, A.E.; Roperch, J.P.; Sayan, B.S.; Rossi, M.; Pinkoski, M.J.; Knight, R.A.; Willis, A.E.; Melino, G. Generation of DeltaTAp73 proteins by translation from a putative internal ribosome entry site. Ann. N. Y. Acad. Sci. 2007, 1095, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Sharathchandra, A.; Katoch, A.; Das, S. IRES mediated translational regulation of p53 isoforms. Wiley Interdiscip. Rev. RNA 2013, 5, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Kim, W.; Lee, K.H.; Kim, S.H.; Lee, H.R.; Kim, H.J.; Jung, Y.; Choi, J.H.; Kim, K.T. hnRNP Q regulates translation of p53 in normal and stress conditions. Cell Death Differ. 2013, 20, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Grover, R.; Ray, P.S.; Das, S. Polypyrimidine tract binding protein regulates IRES-mediated translation of p53 isoforms. Cell Cycle 2008, 7, 2189–2198. [Google Scholar] [CrossRef] [PubMed]
- Montanaro, L.; Calienni, M.; Bertoni, S.; Rocchi, L.; Sansone, P.; Storci, G.; Santini, D.; Ceccarelli, C.; Taffurelli, M.; Carnicelli, D.; et al. Novel dyskerin-mediated mechanism of p53 inactivation through defective mRNA translation. Cancer Res. 2010, 70, 4767–4777. [Google Scholar] [CrossRef] [PubMed]
- Bellodi, C.; Kopmar, N.; Ruggero, D. Deregulation of oncogene-induced senescence and p53 translational control in X-linked dyskeratosis congenita. Embo J. 2010, 29, 1865–1876. [Google Scholar] [CrossRef] [PubMed]
- Sharathchandra, A.; Lal, R.; Khan, D.; Das, S. Annexin A2 and PSF proteins interact with p53 IRES and regulate translation of p53 mRNA. RNA Biol. 2012, 9, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Weingarten-Gabbay, S.; Khan, D.; Liberman, N.; Yoffe, Y.; Bialik, S.; Das, S.; Oren, M.; Kimchi, A. The translation initiation factor DAP5 promotes IRES-driven translation of p53 mRNA. Oncogene 2013, 33, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.; Katoch, A.; Das, A.; Sharathchandra, A.; Lal, R.; Roy, P.; Das, S. Reversible induction of translational isoforms of p53 in glucose deprivation. Cell Death Differ. 2015, 22, 1203–1218. [Google Scholar] [CrossRef] [PubMed]
- Hellen, C.U.; Sarnow, P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 2001, 15, 1593–1612. [Google Scholar] [CrossRef] [PubMed]
- Stoneley, M.; Willis, A.E. Cellular internal ribosome entry segments: Structures, trans-acting factors and regulation of gene expression. Oncogene 2004, 23, 3200–3207. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005, 123, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Fierro-Monti, I.; Mathews, M.B. Proteins binding to duplexed RNA: One motif, multiple functions. Trends Biochem. Sci. 2000, 25, 241–246. [Google Scholar] [CrossRef]
- Xu, Y.H.; Grabowski, G.A. Molecular cloning and characterization of a translational inhibitory protein that binds to coding sequences of the human acid beta-glucosidase and other mRNAs. Mol. Genet. Metab. 1999, 68, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Hartman, T.R.; Qian, S.; Bolinger, C.; Fernandez, S.; Schoenberg, D.R.; Boris-Lawrie, K. RNA helicase A is necessary for translation of selected messenger RNAs. Nat. Struct. Mol. Biol. 2006, 13, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Wedeken, L.; Singh, P.; Klempnauer, K.H. Tumor suppressor protein Pdcd4 inhibits translation of p53 mRNA. J. Biol. Chem. 2011, 286, 42855–42862. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Xu, T.; Ganapathy, S.; Shadfan, M.; Long, M.; Huang, T.H.; Thompson, I.; Yuan, Z.M. Elevated snoRNA biogenesis is essential in breast cancer. Oncogene 2014, 33, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Ma, W.; Benchimol, S. A translation repressor element resides in the 3′ untranslated region of human p53 mRNA. Oncogene 1999, 18, 6419–6424. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kastan, M.B. 5′-3′-UTR interactions regulate p53 mRNA translation and provide a target for modulating p53 induction after DNA damage. Genes Dev. 2010, 24, 2146–2156. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, B.; Hanazawa, M.; Lee, M.H.; Nayak, S.; Volkmann, K.; Hofmann, R.; Hengartner, M.; Schedl, T.; Gartner, A. Translational repression of C. elegans p53 by GLD-1 regulates DNA damage-induced apoptosis. Cell 2005, 120, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Koh, D.C.; Wong, S.M.; Liu, D.X. Synergism of the 3′-untranslated region and an internal ribosome entry site differentially enhances the translation of a plant virus coat protein. J. Biol. Chem. 2003, 278, 20565–20573. [Google Scholar] [CrossRef] [PubMed]
- Gasco, M.; Shami, S.; Crook, T. The p53 pathway in breast cancer. Breast Cancer Res. 2002, 4, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacGrogan, D.; Bookstein, R. Tumour suppressor genes in prostate cancer. Semin. Cancer Biol. 1997, 8, 11–19. [Google Scholar] [CrossRef] [PubMed]
- MacInnes, A.W.; Amsterdam, A.; Whittaker, C.A.; Hopkins, N.; Lees, J.A. Loss of p53 synthesis in zebrafish tumors with ribosomal protein gene mutations. Proc. Natl. Acad. Sci. USA 2008, 105, 10408–10413. [Google Scholar] [CrossRef] [PubMed]
- Montanaro, L.; Trere, D.; Derenzini, M. Nucleolus, ribosomes, and cancer. Am. J. Pathol. 2008, 173, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Yoon, A.; Peng, G.; Brandenburger, Y.; Zollo, O.; Xu, W.; Rego, E.; Ruggero, D. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science 2006, 312, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Jackson, N.L.; Choi, H.; King, P.H.; Emanuel, P.D.; Blume, S.W. Alterations in RNA-binding activities of IRES-regulatory proteins as a mechanism for physiological variability and pathological dysregulation of IGF-IR translational control in human breast tumor cells. J. Cell Physiol. 2008, 217, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, J.R.; Parganas, E.; Lee, Y.; Yang, C.; Wang, J.; Brennan, J.; MacLean, K.H.; Han, J.; Chittenden, T.; Ihle, J.N.; et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 2003, 4, 321–328. [Google Scholar] [CrossRef]
- Garrison, S.P.; Phillips, D.C.; Jeffers, J.R.; Chipuk, J.E.; Parsons, M.J.; Rehg, J.E.; Opferman, J.T.; Green, D.R.; Zambetti, G.P. Genetically defining the mechanism of Puma- and Bim-induced apoptosis. Cell Death Differ. 2012, 19, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Eki, T.; Okumura, K.; Nogami, M.; Soares Vda, C.; Murakami, Y.; Hanaoka, F.; Hurwitz, J. The human RNA helicase A (DDX9) gene maps to the prostate cancer susceptibility locus at chromosome band 1q25 and its pseudogene (DDX9P) to 13q22, respectively. Somat. Cell Mol. Genet. 1999, 25, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hemmerich, P.; Grosse, F. Werner syndrome helicase (WRN), nuclear DNA helicase II (NDH II) and histone gammaH2AX are localized to the centrosome. Cell Biol. Int. 2007, 31, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Schlott, B.; Gorlach, M.; Grosse, F. DNA-dependent protein kinase (DNA-PK) phosphorylates nuclear DNA helicase II/RNA helicase A and hnRNP proteins in an RNA-dependent manner. Nucleic Acids Res. 2004, 32, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Neplioueva, V.; Dobrikova, E.Y.; Mukherjee, N.; Keene, J.D.; Gromeier, M. Tissue type-specific expression of the dsRNA-binding protein 76 and genome-wide elucidation of its target mRNAs. PLoS ONE 2010, 5, e11710. [Google Scholar] [CrossRef] [PubMed]
- Van Maerken, T.; Rihani, A.; Van Goethem, A.; De Paepe, A.; Speleman, F.; Vandesompele, J. Pharmacologic activation of wild-type p53 by nutlin therapy in childhood cancer. Cancer Lett. 2013, 344, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Tovar, C.; Rosinski, J.; Filipovic, Z.; Higgins, B.; Kolinsky, K.; Hilton, H.; Zhao, X.; Vu, B.T.; Qing, W.; Packman, K.; et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: Implications for therapy. Proc. Natl. Acad. Sci. USA 2006, 103, 1888–1893. [Google Scholar] [CrossRef] [PubMed]
- Barone, G.; Tweddle, D.A.; Shohet, J.M.; Chesler, L.; Moreno, L.; Pearson, A.D.; Van Maerken, T. MDM2-p53 interaction in paediatric solid tumours: Preclinical rationale, biomarkers and resistance. Curr. Drug Targets 2014, 15, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.H.; Shen, H.; Maki, C.G. Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene 2011, 30, 4678–4686. [Google Scholar] [CrossRef] [PubMed]
- Saiki, A.Y.; Caenepeel, S.; Cosgrove, E.; Su, C.; Boedigheimer, M.; Oliner, J.D. Identifying the determinants of response to MDM2 inhibition. Oncotarget 2015, 6, 7701–7712. [Google Scholar] [CrossRef] [PubMed]
- Stegh, A.H. Targeting the p53 signaling pathway in cancer therapy—The promises, challenges and perils. Expert Opin. Ther. Targets 2012, 16, 67–83. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, A.; Graff, J.R. eIF-4E expression and its role in malignancies and metastases. Oncogene 2004, 23, 3189–3199. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, B.; Benyumov, A.O.; Ghosh, P.; Jia, Y.; Avdulov, S.; Dahlberg, P.S.; Peterson, M.; Smith, K.; Polunovsky, V.A.; Bitterman, P.B.; et al. Nontoxic chemical interdiction of the epithelial-to-mesenchymal transition by targeting cap-dependent translation. ACS Chem. Biol. 2009, 4, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Polunovsky, V.; Bitterman, P.B.; Wagner, C.R. Cap-dependent translation initiation factor eIF4E: An emerging anticancer drug target. Med. Res. Rev. 2012, 32, 786–814. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jia, Y.; Jacobson, B.; McCauley, J.; Kratzke, R.; Bitterman, P.B.; Wagner, C.R. Treatment of breast and lung cancer cells with a N-7 benzyl guanosine monophosphate tryptamine phosphoramidate pronucleotide (4Ei-1) results in chemosensitization to gemcitabine and induced eIF4E proteasomal degradation. Mol. Pharm. 2013, 10, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Moerke, N.J.; Aktas, H.; Chen, H.; Cantel, S.; Reibarkh, M.Y.; Fahmy, A.; Gross, J.D.; Degterev, A.; Yuan, J.; Chorev, M.; et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 2007, 128, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Aktas, B.H.; Wang, Y.; He, X.; Sahoo, R.; Zhang, N.; Denoyelle, S.; Kabha, E.; Yang, H.; Freedman, R.Y.; et al. Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget 2012, 3, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Kabha, E.; Papadopoulos, E.; Wagner, G. 4EGI-1 targets breast cancer stem cells by selective inhibition of translation that persists in CSC maintenance, proliferation and metastasis. Oncotarget 2014, 5, 6028–6037. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, E.; Jenni, S.; Kabha, E.; Takrouri, K.J.; Yi, T.; Salvi, N.; Luna, R.E.; Gavathiotis, E.; Mahalingam, P.; Arthanari, H.; et al. Structure of the eukaryotic translation initiation factor eIF4E in complex with 4EGI-1 reveals an allosteric mechanism for dissociating eIF4G. Proc. Natl. Acad. Sci. USA 2014, 111, E3187–E3195. [Google Scholar] [CrossRef] [PubMed]
- Avdulov, S.; Li, S.; Michalek, V.; Burrichter, D.; Peterson, M.; Perlman, D.M.; Manivel, J.C.; Sonenberg, N.; Yee, D.; Bitterman, P.B.; Polunovsky, V.A. Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell 2004, 5, 553–563. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, B.; Harris, B.R.E.; Liu, Y.; Deng, Y.; Gradilone, S.A.; Cleary, M.P.; Liu, J.; Yang, D.-Q. Targeting IRES-Mediated p53 Synthesis for Cancer Diagnosis and Therapeutics. Int. J. Mol. Sci. 2017, 18, 93. https://doi.org/10.3390/ijms18010093
Ji B, Harris BRE, Liu Y, Deng Y, Gradilone SA, Cleary MP, Liu J, Yang D-Q. Targeting IRES-Mediated p53 Synthesis for Cancer Diagnosis and Therapeutics. International Journal of Molecular Sciences. 2017; 18(1):93. https://doi.org/10.3390/ijms18010093
Chicago/Turabian StyleJi, Bai, Benjamin R. E. Harris, Yahui Liu, Yibin Deng, Sergio A. Gradilone, Margot P. Cleary, Jianhua Liu, and Da-Qing Yang. 2017. "Targeting IRES-Mediated p53 Synthesis for Cancer Diagnosis and Therapeutics" International Journal of Molecular Sciences 18, no. 1: 93. https://doi.org/10.3390/ijms18010093
APA StyleJi, B., Harris, B. R. E., Liu, Y., Deng, Y., Gradilone, S. A., Cleary, M. P., Liu, J., & Yang, D. -Q. (2017). Targeting IRES-Mediated p53 Synthesis for Cancer Diagnosis and Therapeutics. International Journal of Molecular Sciences, 18(1), 93. https://doi.org/10.3390/ijms18010093