
Recognition of Local DNA Structures by p53 Protein


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Local DNA Structures
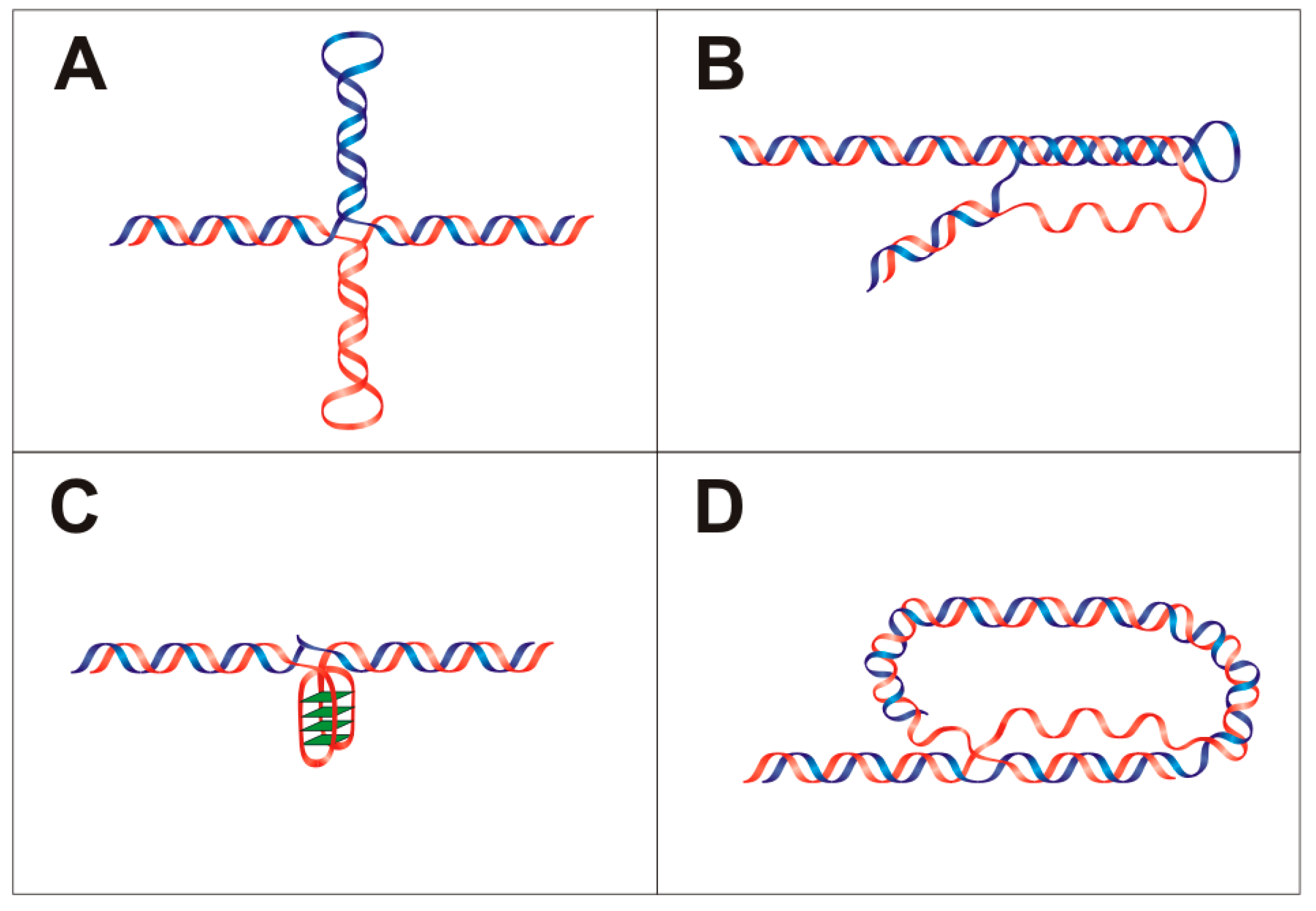
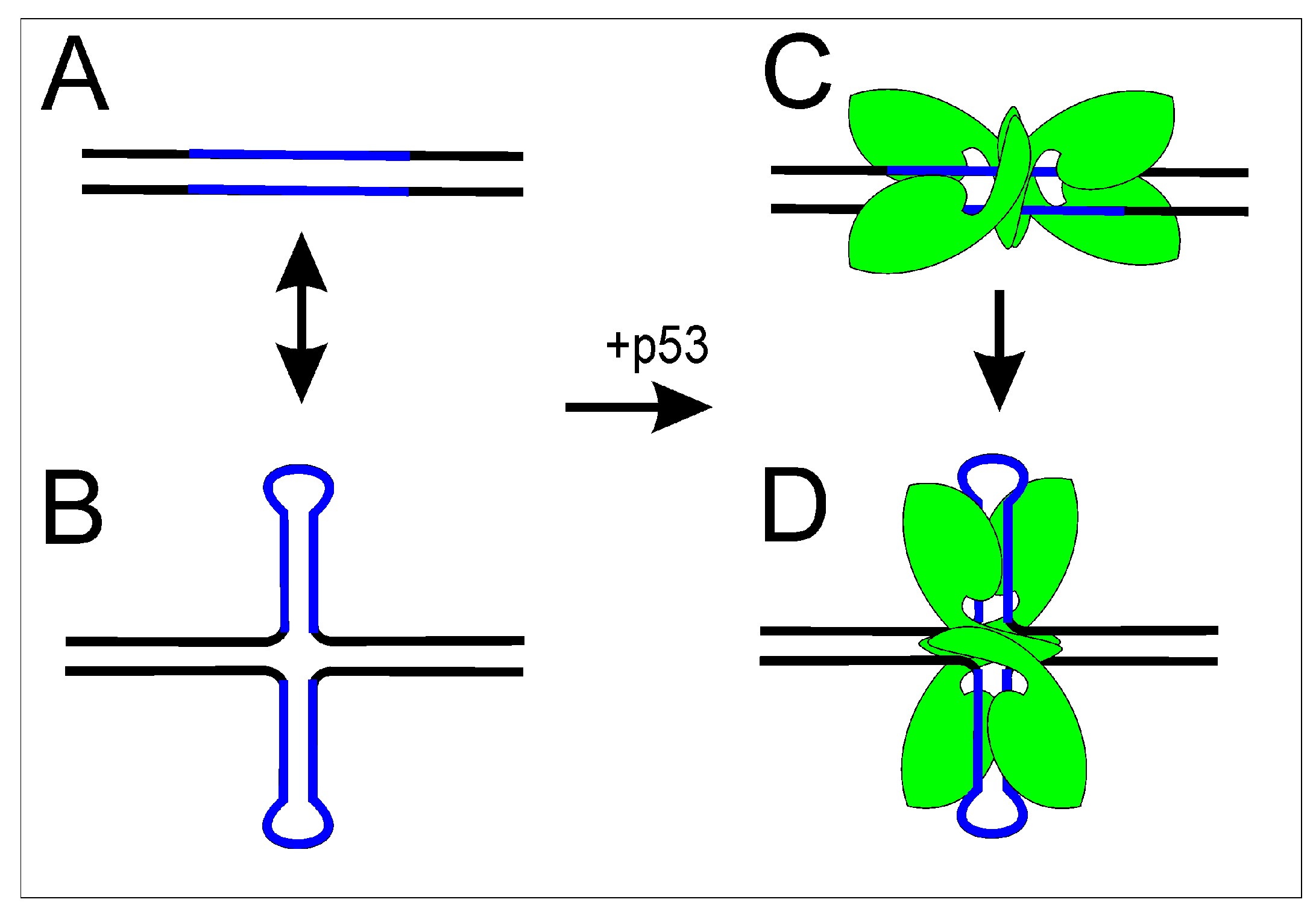
1.1.1. Hairpins and Cruciform Structures
1.1.2. Triplexes
1.1.3. Quadruplexes
1.1.4. T-Loops
2. Interaction of p53 with DNA
2.1. Sequence-Specific Interaction
2.1.1. DNA Bending by p53 Protein Binding
2.1.2. p53 Binding to p53 Target Site Enhanced by Cruciform Extrusion
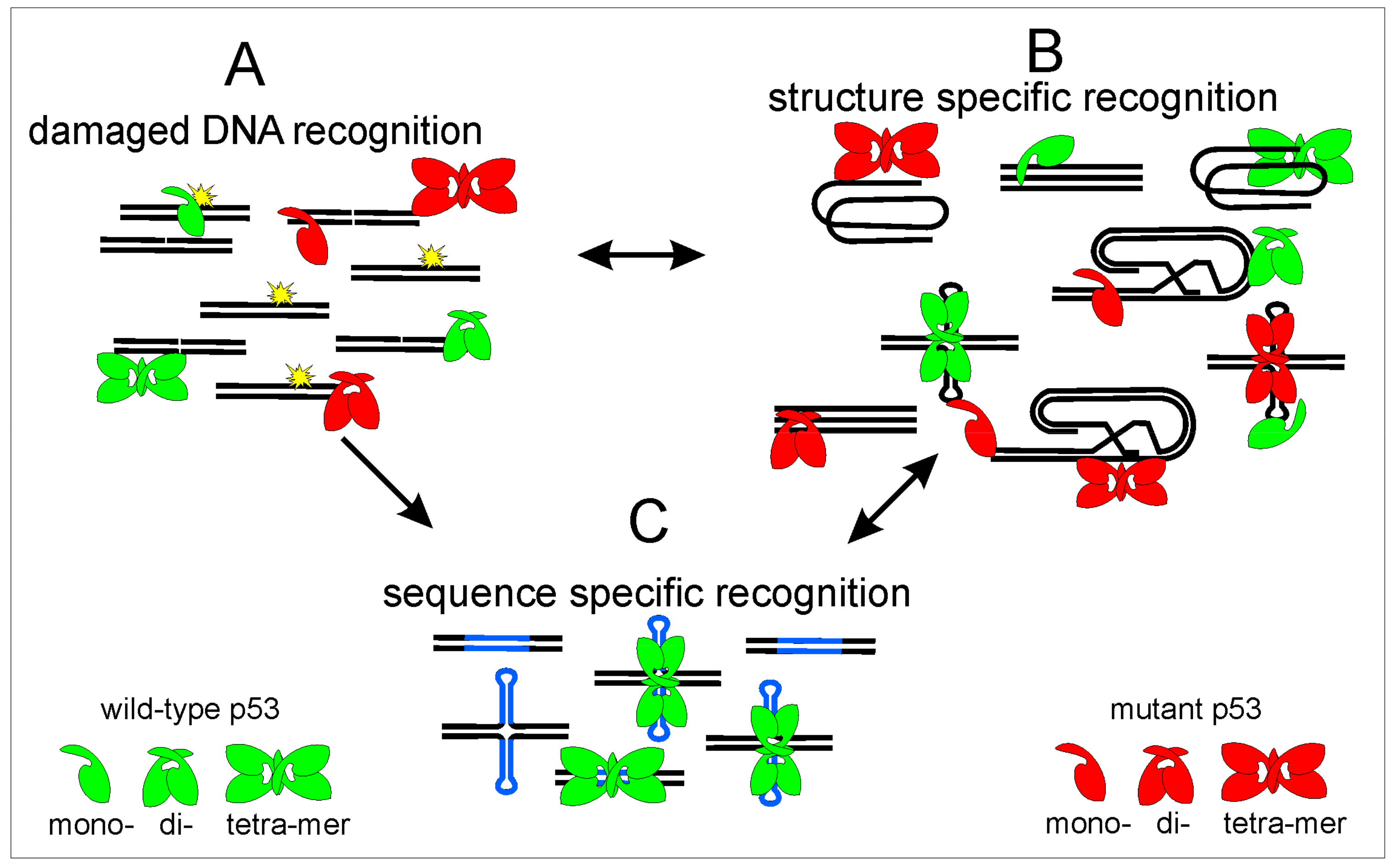
2.2. p53 Binding to Local DNA Structures
2.2.1. Bulged DNA and Mismatches
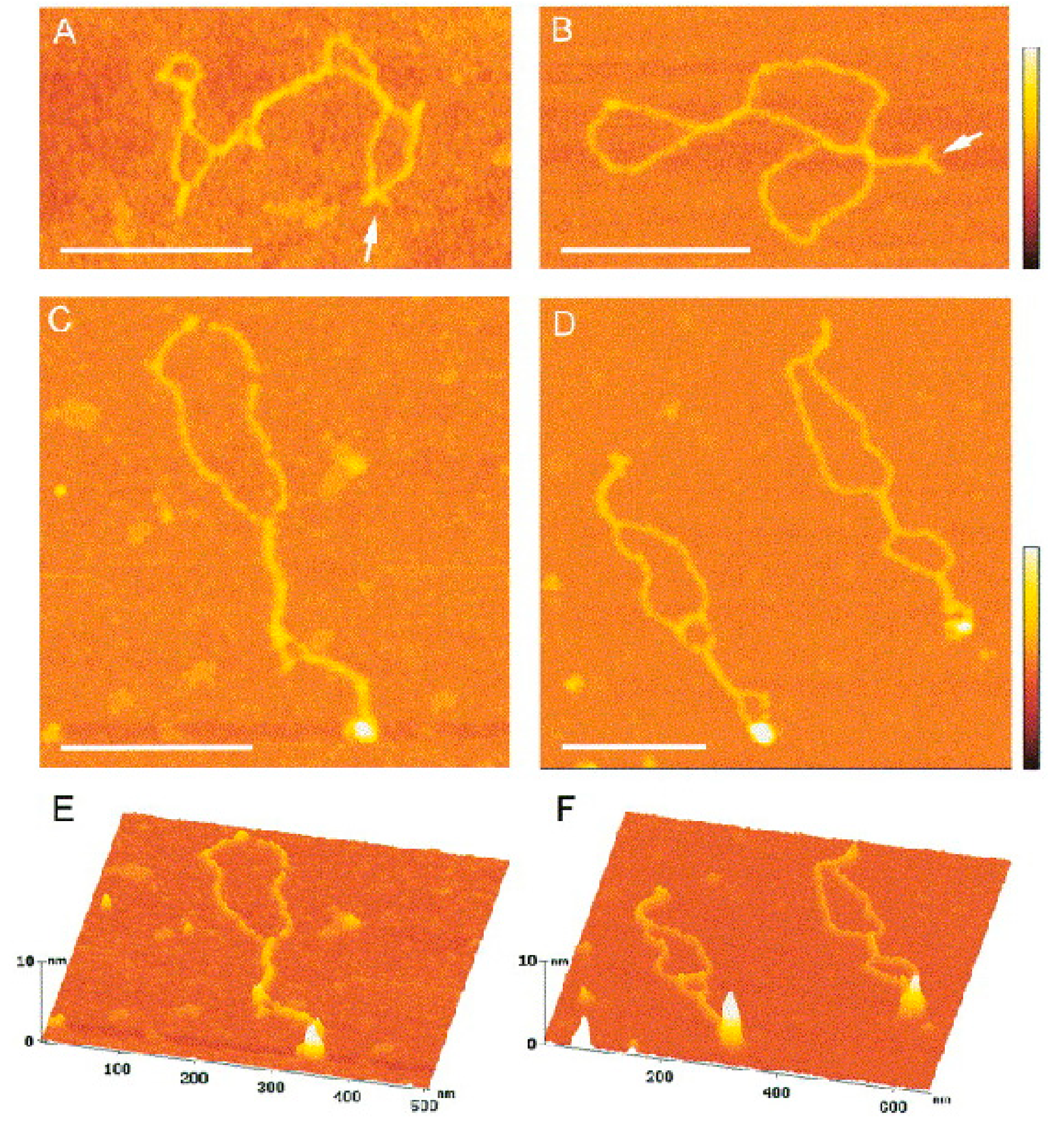
2.2.2. Holliday Junctions and Cruciforms
2.2.3. Hemicatenane DNA
2.2.4. Telomeric T-Loop and Single Strand Overhangs
2.2.5. Triplexes
2.2.6. Quadruplexes
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Goh, A.M.; Coffill, C.R.; Lane, D.P. The role of mutant p53 in human cancer. J. Pathol. 2011, 223, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W. Regulation of the p53 response and its relationship to cancer. Biochem. J. 2015, 469, 325–346. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Lohrum, M.A.; Vousden, K.H. Regulation and activation of p53 and its family members. Cell Death Differ. 1999, 6, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, X.Q.; Du, J.Z. Cellular adaptation to hypoxia and p53 transcription regulation. J. Zhejiang Univ. Sci. B 2009, 10, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Murata, A.; Sakamoto, S.; Nanatani, K.; Wada, T.; Takahashi, S.; Kamagata, K. Activation of p53 facilitates the target search in DNA by enhancing the target recognition probability. J. Mol. Biol. 2016, 428, 2916–2930. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Majima, T. Conformational changes of non-B DNA. Chem. Soc. Rev. 2011, 40, 5893–5909. [Google Scholar] [CrossRef] [PubMed]
- Palecek, E. Local supercoil-stabilized DNA structures. Crit. Rev. Biochem. Mol. Biol. 1991, 26, 151–226. [Google Scholar] [CrossRef] [PubMed]
- Van Holde, K.; Zlatanova, J. Unusual DNA structures, chromatin and transcription. Bioessays 1994, 16, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.D. Non-B DNA conformations, mutagenesis and disease. Trends Biochem. Sci. 2007, 32, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Cer, R.Z.; Bruce, K.H.; Donohue, D.E.; Temiz, N.A.; Mudunuri, U.S.; Yi, M.; Volfovsky, N.; Bacolla, A.; Luke, B.T.; Collins, J.R.; et al. Searching for non-B DNA-forming motifs using nBMST (non-B DNA motif search tool). In Current Protocols in Human Genetics; John Wiley Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 1–22. [Google Scholar]
- Chasovskikh, S.; Dimtchev, A.; Smulson, M.; Dritschilo, A. DNA transitions induced by binding of PARP-1 to cruciform structures in supercoiled plasmids. Cytometry A 2005, 68, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Limanskaia, O. Species-specific detection of Mycobacterium tuberculosis complex. Probl. Tuberk. Bolezn. Legk. 2009, 10, 49–55. [Google Scholar]
- Mikheikin, A.L.; Lushnikov, A.Y.; Lyubchenko, Y.L. Effect of DNA supercoiling on the geometry of holliday junctions. Biochemistry 2006, 45, 12998–13006. [Google Scholar] [CrossRef] [PubMed]
- Pearson, C.E.; Zorbas, H.; Price, G.B.; Zannis-Hadjopoulos, M. Inverted repeats, stem-loops, and cruciforms: Significance for initiation of DNA replication. J. Cell. Biochem. 1996, 63, 1–22. [Google Scholar] [CrossRef]
- Werbowy, K.; Cieslinski, H.; Kur, J. Characterization of a cryptic plasmid pSFKW33 from Shewanella sp. 33B. Plasmid 2009, 62, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Brazda, V.; Kolomaznik, J.; Lysek, J.; Haronikova, L.; Coufal, J.; St’astny, J. Palindrome analyser—A new web-based server for predicting and evaluating inverted repeats in nucleotide sequences. Biochem. Biophys. Res. Commun. 2016, 478, 1739–1745. [Google Scholar] [CrossRef] [PubMed]
- Cer, R.Z.; Bruce, K.H.; Mudunuri, U.S.; Yi, M.; Volfovsky, N.; Luke, B.T.; Bacolla, A.; Collins, J.R.; Stephens, R.M. Non-B DB: A database of predicted non-B DNA-forming motifs in mammalian genomes. Nucleic Acids Res. 2011, 39, D383–D391. [Google Scholar] [CrossRef] [PubMed]
- Aranda, A.; Perez-Ortin, J.E.; Benham, C.J.; Del Olmo, M.L. Analysis of the structure of a natural alternating d(TA)n sequence in yeast chromatin. Yeast 1997, 13, 313–326. [Google Scholar] [CrossRef]
- Kurahashi, H.; Inagaki, H.; Yamada, K.; Ohye, T.; Taniguchi, M.; Emanuel, B.S.; Toda, T. Cruciform DNA structure underlies the etiology for palindrome-mediated human chromosomal translocations. J. Biol. Chem. 2004, 279, 35377–35383. [Google Scholar] [CrossRef] [PubMed]
- Lyubchenko, Y.L. DNA structure and dynamics: An atomic force microscopy study. Cell Biochem. Biophys. 2004, 41, 75–98. [Google Scholar] [CrossRef]
- Shlyakhtenko, L.S.; Potaman, V.N.; Sinden, R.R.; Lyubchenko, Y.L. Structure and dynamics of supercoil-stabilized DNA cruciforms. J. Mol. Biol. 1998, 280, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Shlyakhtenko, L.S.; Hsieh, P.; Grigoriev, M.; Potaman, V.N.; Sinden, R.R.; Lyubchenko, Y.L. A cruciform structural transition provides a molecular switch for chromosome structure and dynamics. J. Mol. Biol. 2000, 296, 1169–1173. [Google Scholar] [CrossRef] [PubMed]
- Panayotatos, N.; Fontaine, A. A native cruciform DNA structure probed in bacteria by recombinant T7 endonuclease. J. Biol. Chem. 1987, 262, 11364–11368. [Google Scholar] [PubMed]
- Yamaguchi, K.; Yamaguchi, M. The replication origin of pSC101: The nucleotide sequence and replication functions of the ori region. Gene 1984, 29, 211–219. [Google Scholar] [PubMed]
- Brazda, V.; Laister, R.C.; Jagelska, E.B.; Arrowsmith, C. Cruciform structures are a common DNA feature important for regulating biological processes. BMC Mol. Biol. 2011, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Manelyte, L.; Strohner, R.; Gross, T.; Langst, G. Chromatin targeting signals, nucleosome positioning mechanism and non-coding RNA-mediated regulation of the chromatin remodeling complex NoRC. PLoS Genet. 2014, 10, e1004157. [Google Scholar] [CrossRef] [PubMed]
- Yahyaoui, W.; Callejo, M.; Price, G.B.; Zannis-Hadjopoulos, M. Deletion of the cruciform binding domain in CBP/14-3-3 displays reduced origin binding and initiation of DNA replication in budding yeast. BMC Mol. Biol. 2007, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Marins, M.; Kamisugi, Y.; Meyer, P. Analysis of hypermethylation in the RPS element suggests a signal function for short inverted repeats in de novo methylation. Plant Mol. Biol. 2002, 48, 383–399. [Google Scholar] [CrossRef] [PubMed]
- Gentry, M.; Hennig, L. A structural bisulfite assay to identify DNA cruciforms. Mol. Plant 2016, 9, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Frank-Kamenetskii, M.D.; Mirkin, S.M. Triplex DNA structures. Annu. Rev. Biochem. 1995, 64, 65–95. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Vasquez, K.M. Triplex technology in studies of DNA damage, DNA repair, and mutagenesis. Biochimie 2011, 93, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Khutsishvili, I.; Marky, L.A. DNA complexes containing joined triplex and duplex motifs: Melting behavior of intramolecular and bimolecular complexes with similar sequences. J. Phys. Chem. B 2010, 114, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Wells, R.D. Non-B DNA conformations as determinants of mutagenesis and human disease. Mol. Carcinog. 2009, 48, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Schroth, G.P.; Ho, P.S. Occurrence of potential cruciform and H-DNA forming sequences in genomic DNA. Nucleic Acids Res. 1995, 23, 1977–1983. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Gaddis, S.S.; MacLeod, M.C.; Walborg, E.F.; Thames, H.D.; di Giovanni, J.; Vasquez, K.M. High-affinity triplex-forming oligonucleotide target sequences in mammalian genomes. Mol. Carcinog. 2007, 46, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Collins, J.R.; Gold, B.; Chuzhanova, N.; Yi, M.; Stephens, R.M.; Stefanov, S.; Olsh, A.; Jakupciak, J.P.; Dean, M.; et al. Long homopurine·homopyrimidine sequences are characteristic of genes expressed in brain and the pseudoautosomal region. Nucleic Acids Res. 2006, 34, 2663–2675. [Google Scholar] [CrossRef] [PubMed]
- Gorab, E.; Amabis, J.M.; Stocker, A.J.; Drummond, L.; Stollar, B.D. Potential sites of triple-helical nucleic acid formation in chromosomes of Rhynchosciara (Diptera: Sciaridae) and Drosophila melanogaster. Chromosome Res. 2009, 17, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Hoyne, P.R.; Maher, L.J., 3rd. Functional studies of potential intrastrand triplex elements in the Escherichia coli genome. J. Mol. Biol. 2002, 318, 373–386. [Google Scholar] [CrossRef]
- Krasilnikova, M.M.; Mirkin, S.M. Replication stalling at Friedreich’s ataxia (GAA)n repeats in vivo. Mol. Cell. Biol. 2004, 24, 2286–2295. [Google Scholar] [CrossRef] [PubMed]
- Mariappan, S.V.; Catasti, P.; Silks, L.A., 3rd; Bradbury, E.M.; Gupta, G. The high-resolution structure of the triplex formed by the GAA/TTC triplet repeat associated with Friedreich’s ataxia. J. Mol. Biol. 1999, 285, 2035–2052. [Google Scholar] [CrossRef] [PubMed]
- Rajeswari, M.R. DNA triplex structures in neurodegenerative disorder, Friedreich’s ataxia. J. Biosci. 2012, 37, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Rajeswari, M.R.; Ahmed, F. Formation and thermodynamic stability of intermolecular (R*R·Y) DNA triplex in GAA/TTC repeats associated with Freidreich’s ataxia. J. Biomol. Struct. Dyn. 2002, 19, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Bowater, R.P.; Wells, R.D. The intrinsically unstable life of DNA triplet repeats associated with human hereditary disorders. Prog. Nucleic Acid. Res. Mol. Biol. 2001, 66, 159–202. [Google Scholar] [PubMed]
- Singh, H.N.; Rajeswari, M.R. Role of long purine stretches in controlling the expression of genes associated with neurological disorders. Gene 2015, 572, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S.; Parkinson, G.N. Quadruplex DNA crystal structures and drug design. Biochimie 2008, 90, 1184–1196. [Google Scholar] [CrossRef] [PubMed]
- Gellert, M.; Lipsett, M.N.; Davies, D.R. Helix formation by guanylic acid. Proc. Natl. Acad. Sci. USA 1962, 48, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Patel, D.J. Solution structure of the human telomeric repeat d[AG3(T2AG3)3] G-tetraplex. Structure 1993, 1, 263–282. [Google Scholar] [CrossRef]
- Huppert, J.L.; Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Kikin, O.; D’Antonio, L.; Bagga, P.S. QGRS Mapper: A web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006, 34, W676–W682. [Google Scholar] [CrossRef] [PubMed]
- Scaria, V.; Hariharan, M.; Arora, A.; Maiti, S. Quadfinder: Server for identification and analysis of quadruplex-forming motifs in nucleotide sequences. Nucleic Acids Res. 2006, 34, W683–W685. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Bugaut, A.; Huppert, J.L.; Balasubramanian, S. An RNA G-quadruplex in the 5′ UTR of the NRAS proto-oncogene modulates translation. Nat. Chem. Biol. 2007, 3, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Schaffitzel, C.; Berger, I.; Postberg, J.; Hanes, J.; Lipps, H.J.; Pluckthun, A. In vitro generated antibodies specific for telomeric guanine-quadruplex DNA react with Stylonychia lemnae macronuclei. Proc. Natl. Acad. Sci. USA 2001, 98, 8572–8577. [Google Scholar] [PubMed]
- Yang, Q.; Xiang, J.; Yang, S.; Zhou, Q.; Li, Q.; Tang, Y.; Xu, G. Verification of specific G-quadruplex structure by using a novel cyanine dye supramolecular assembly: I. recognizing mixed G-quadruplex in human telomeres. Chem. Commun. 2009, 9, 1103–1105. [Google Scholar] [CrossRef] [PubMed]
- De Cian, A.; Gros, J.; Guedin, A.; Haddi, M.; Lyonnais, S.; Guittat, L.; Riou, J.F.; Trentesaux, C.; Sacca, B.; Lacroix, L.; et al. DNA and RNA quadruplex ligands. Nucleic Acids Symp. Ser. 2008, 52, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Eddy, J.; Maizels, N. Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res. 2006, 34, 3887–3896. [Google Scholar] [CrossRef] [PubMed]
- Eddy, J.; Maizels, N. Selection for the G4 DNA motif at the 5’ end of human genes. Mol. Carcinog. 2009, 48, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian telomeres end in a large duplex loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef]
- Stansel, R.M.; de Lange, T.; Griffith, J.D. T-loop assembly in vitro involves binding of TRF2 near the 3′ telomeric overhang. EMBO J. 2001, 20, 5532–5540. [Google Scholar] [CrossRef] [PubMed]
- Murti, K.G.; Prescott, D.M. Telomeres of polytene chromosomes in a ciliated protozoan terminate in duplex DNA loops. Proc. Natl. Acad. Sci. USA 1999, 96, 14436–14439. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Jordan, J.L.; Cross, G.A.; de Lange, T.; Griffith, J.D. T-loops at trypanosome telomeres. EMBO J. 2001, 20, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Yang, Y.; Tantoso, E.; Chua, G.H.; Yeo, Z.X.; Ng, F.S.; Wong, S.T.; Chung, C.W.; Li, K.B. In silico analysis of p53 using the p53 knowledgebase: Mutations, polymorphisms, microRNAs and pathways. In Silico Biol. 2007, 7, 61–75. [Google Scholar] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef] [PubMed]
- Okorokov, A.L.; Orlova, E.V. Structural biology of the p53 tumour suppressor. Curr. Opin. Struct. Biol. 2009, 19, 197–202. [Google Scholar] [CrossRef] [PubMed]
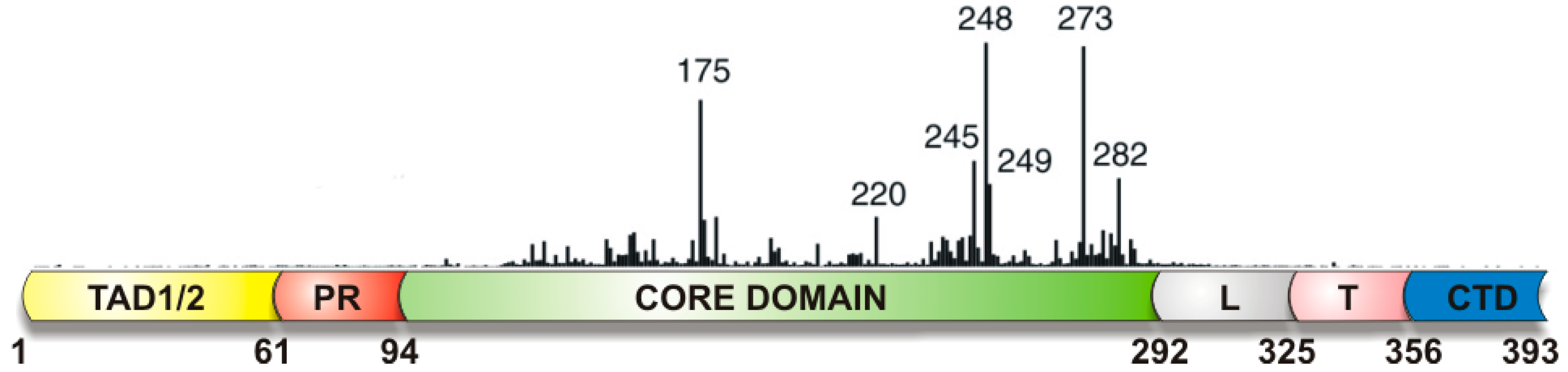
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on Tp53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC Tp53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Wang, T.; Naumovski, L.; Lopez, C.D.; Brachmann, R.K. Groups of p53 target genes involved in specific p53 downstream effects cluster into different classes of DNA binding sites. Oncogene 2002, 21, 7901–7911. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S.; Kern, S.E.; Pietenpol, J.A.; Kinzler, K.W.; Vogelstein, B. Definition of a consensus binding site for p53. Nat. Genet. 1992, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
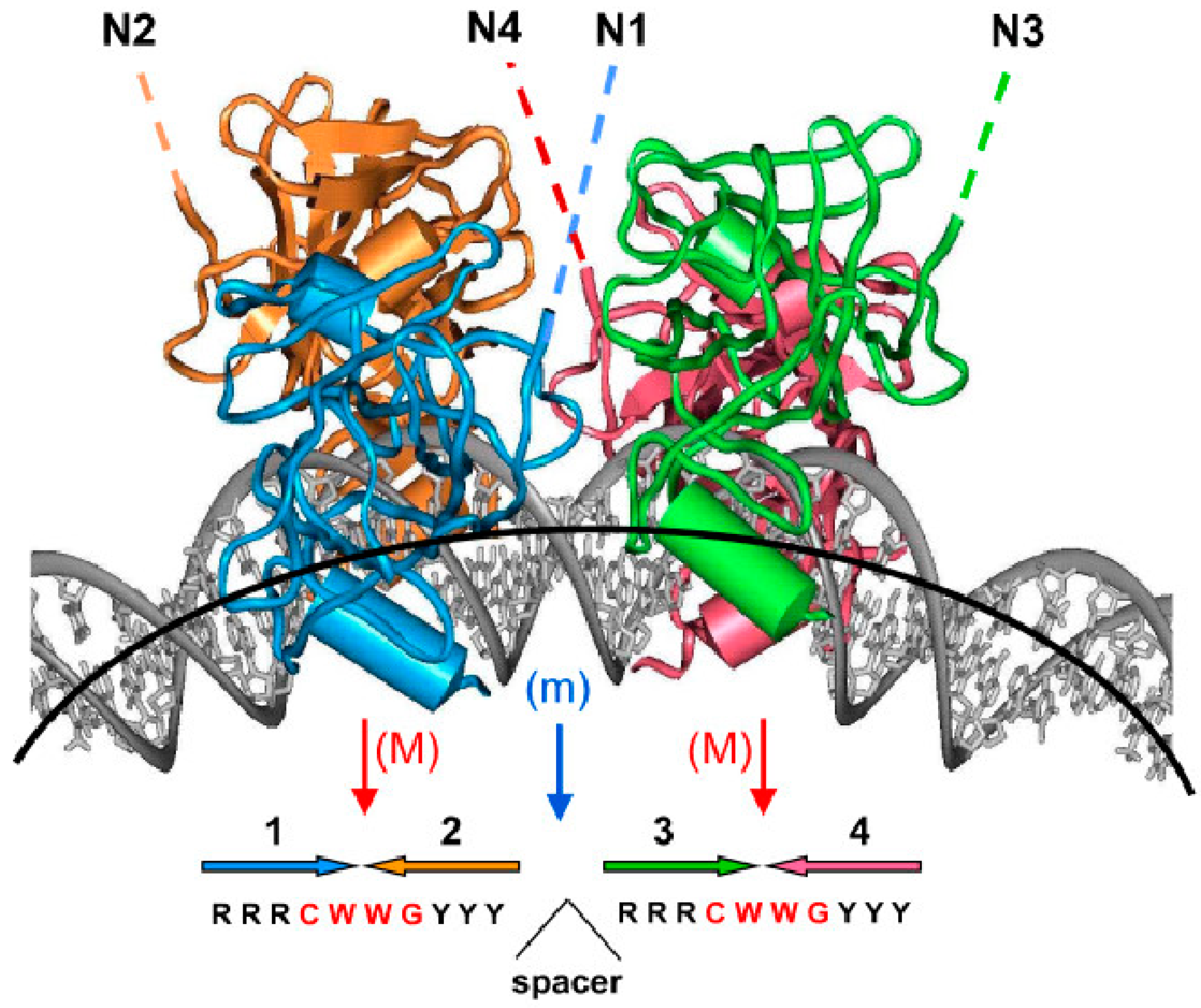
- Balagurumoorthy, P.; Sakamoto, H.; Lewis, M.S.; Zambrano, N.; Clore, G.M.; Gronenborn, A.M.; Appella, E.; Harrington, R.E. Four p53 DNA-binding domain peptides bind natural p53-response elements and bend the DNA. Proc. Natl. Acad. Sci. USA 1995, 92, 8591–8595. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.L.; Veprintsev, D.B.; Bycroft, M.; Fersht, A.R. Comparative binding of p53 to its promoter and DNA recognition elements. J. Mol. Biol. 2005, 348, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Albrechtsen, N.; Deppert, W. DNA-conformation is an important determinant of sequence-specific DNA binding by tumor suppressor p53. Oncogene 1997, 15, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Inga, A.; Storici, F.; Darden, T.A.; Resnick, M.A. Differential transactivation by the p53 transcription factor is highly dependent on p53 level and promoter target sequence. Mol. Cell. Biol. 2002, 22, 8612–8625. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Pan, Y.; Zheng, J.; Levine, A.J.; Nussinov, R. Sequence analysis of p53 response-elements suggests multiple binding modes of the p53 tetramer to DNA targets. Nucleic Acids Res. 2007, 35, 2986–3001. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dey, R.; Chen, L. Crystal structure of the p53 core domain bound to a full consensus site as a self-assembled tetramer. Structure 2010, 18, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Gohler, T.; Reimann, M.; Cherny, D.; Walter, K.; Warnecke, G.; Kim, E.; Deppert, W. Specific interaction of p53 with target binding sites is determined by DNA conformation and is regulated by the C-terminal domain. J. Biol. Chem. 2002, 277, 41192–41203. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.J.; Menendez, D.; Inga, A.; Noureddine, M.; Bell, D.A.; Resnick, M.A. Noncanonical DNA motifs as transactivation targets by wild type and mutant p53. PLoS Genet. 2008, 4, e1000104. [Google Scholar] [CrossRef]
- McKinney, K.; Prives, C. Efficient specific DNA binding by p53 requires both its central and C-terminal domains as revealed by studies with high-mobility group 1 protein. Mol. Cell. Biol. 2002, 22, 6797–6808. [Google Scholar] [CrossRef] [PubMed]
- McKinney, K.; Mattia, M.; Gottifredi, V.; Prives, C. p53 linear diffusion along DNA requires its C terminus. Mol. Cell 2004, 16, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Brazda, V.; Jagelska, E.B.; Fojta, M.; Palecek, E. Searching for target sequences by p53 protein is influenced by DNA length. Biochem. Biophys. Res. Commun. 2006, 341, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Murata, A.; Ito, Y.; Kashima, R.; Kanbayashi, S.; Nanatani, K.; Igarashi, C.; Okumura, M.; Inaba, K.; Tokino, T.; Takahashi, S.; et al. One-dimensional sliding of p53 along DNA is accelerated in the presence of Ca2+ or Mg2+ at millimolar concentrations. J. Mol. Biol. 2015, 427, 2663–2678. [Google Scholar] [CrossRef] [PubMed]
- Tafvizi, A.; Huang, F.; Fersht, A.R.; Mirny, L.A.; van Oijen, A.M. A single-molecule characterization of p53 search on DNA. Proc. Natl. Acad. Sci. USA 2011, 108, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Leith, J.S.; Tafvizi, A.; Huang, F.; Uspal, W.E.; Doyle, P.S.; Fersht, A.R.; Mirny, L.A.; van Oijen, A.M. Sequence-dependent sliding kinetics of p53. Proc. Natl. Acad. Sci. USA 2012, 109, 16552–16557. [Google Scholar] [CrossRef] [PubMed]
- Terakawa, T.; Kenzaki, H.; Takada, S. p53 searches on DNA by rotation-uncoupled sliding at C-terminal tails and restricted hopping of core domains. J. Am. Chem. Soc. 2012, 134, 14555–14562. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.; Weng, Z.; et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006, 124, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Veprintsev, D.B.; Fersht, A.R. Algorithm for prediction of tumour suppressor p53 affinity for binding sites in DNA. Nucleic Acids Res. 2008, 36, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Inga, A.; Resnick, M.A. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xiao, Z.; Ren, E.C. Redefining the p53 response element. Proc. Natl. Acad. Sci. USA 2009, 106, 14373–14378. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.A.; Andrysik, Z.; Dengler, V.L.; Mellert, H.S.; Guarnieri, A.; Freeman, J.A.; Sullivan, K.D.; Galbraith, M.D.; Luo, X.; Kraus, W.L.; et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. eLife 2014, 3, e02200. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.S.; Chen, X.A.; Park, B.; Rhee, H.S.; Li, P.; Han, K.H.; Mishra, T.; Chan-Salis, K.Y.; Li, Y.; Hardison, R.C.; et al. A comprehensive and high-resolution genome-wide response of p53 to stress. Cell Rep. 2014, 8, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Nagaich, A.K.; Appella, E.; Harrington, R.E. DNA bending is essential for the site-specific recognition of DNA response elements by the DNA binding domain of the tumor suppressor protein p53. J. Biol. Chem. 1997, 272, 14842–14849. [Google Scholar] [CrossRef] [PubMed]
- Nagaich, A.K.; Zhurkin, V.B.; Durell, S.R.; Jernigan, R.L.; Appella, E.; Harrington, R.E. p53-induced DNA bending and twisting: p53 tetramer binds on the outer side of a DNA loop and increases DNA twisting. Proc. Natl. Acad. Sci. USA 1999, 96, 1875–1880. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.J.; Menendez, D.; Sharav, J.; Beno, I.; Rosenthal, K.; Resnick, M.A.; Haran, T.E. Low-level p53 expression changes transactivation rules and reveals superactivating sequences. Proc. Natl. Acad. Sci. USA 2012, 109, 14387–14392. [Google Scholar] [CrossRef] [PubMed]
- Petty, T.J.; Emamzadah, S.; Costantino, L.; Petkova, I.; Stavridi, E.S.; Saven, J.G.; Vauthey, E.; Halazonetis, T.D. An induced fit mechanism regulates p53 DNA binding kinetics to confer sequence specificity. EMBO J. 2011, 30, 2167–2176. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, J.L.; Tainer, J.A. p53 conformational switching for selectivity may reveal a general solution for specific DNA binding. EMBO J. 2011, 30, 2099–2100. [Google Scholar] [CrossRef] [PubMed]
- Demir, O.; Ieong, P.U.; Amaro, R.E. Full-length p53 tetramer bound to DNA and its quaternary dynamics. Oncogene 2016. [Google Scholar] [CrossRef] [PubMed]
- Niederweis, M.; Hillen, W. Electrophoretic analysis of protein-induced DNA bending and twist changes. Electrophoresis 1993, 14, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Palecek, E.; Vlk, D.; Stankova, V.; Brazda, V.; Vojtesek, B.; Hupp, T.R.; Schaper, A.; Jovin, T.M. Tumor suppressor protein p53 binds preferentially to supercoiled DNA. Oncogene 1997, 15, 2201–2209. [Google Scholar] [CrossRef] [PubMed]
- Brazdova, M.; Navratilova, L.; Tichy, V.; Nemcova, K.; Lexa, M.; Hrstka, R.; Pecinka, P.; Adamik, M.; Vojtesek, B.; Palecek, E.; et al. Preferential binding of hot spot mutant p53 proteins to supercoiled DNA in vitro and in cells. PLoS ONE 2013, 8, e59567. [Google Scholar] [CrossRef] [PubMed]
- Adamik, M.; Kejnovska, I.; Bazantova, P.; Petr, M.; Renciuk, D.; Vorlickova, M.; Brazdova, M. p53 binds human telomeric G-quadruplex in vitro. Biochimie 2016, 128, 83–91. [Google Scholar] [CrossRef]
- Kim, E.; Rohaly, G.; Heinrichs, S.; Gimnopoulos, D.; Meissner, H.; Deppert, W. Influence of promoter DNA topology on sequence-specific DNA binding and transactivation by tumor suppressor p53. Oncogene 1999, 18, 7310–7318. [Google Scholar] [CrossRef] [PubMed]
- Jagelska, E.B.; Brazda, V.; Pecinka, P.; Palecek, E.; Fojta, M. DNA topology influences p53 sequence-specific DNA binding through structural transitions within the target sites. Biochem. J. 2008, 412, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Palecek, E.; Brazda, V.; Jagelska, E.; Pecinka, P.; Karlovska, L.; Brazdova, M. Enhancement of p53 sequence-specific binding by DNA supercoiling. Oncogene 2004, 23, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- Jagelska, E.B.; Pivonkova, H.; Fojta, M.; Brazda, V. The potential of the cruciform structure formation as an important factor influencing p53 sequence-specific binding to natural DNA targets. Biochem. Biophys. Res. Commun. 2010, 391, 1409–1414. [Google Scholar] [CrossRef] [PubMed]
- Coufal, J.; Jagelska, E.B.; Liao, J.C.; Brazda, V. Preferential binding of p53 tumor suppressor to p21 promoter sites that contain inverted repeats capable of forming cruciform structure. Biochem. Biophys. Res. Commun. 2013, 441, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Brázda, V.; Čechová, J.; Battistin, M.; Coufal, J.; Jagelská, E.B.; Raimondi, I.; Inga, A. The structure formed by inverted repeats in p53 response elements determines the transactivation activity of p53 protein. Biochem. Biophys. Res. Commun. 2017, 483, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Degtyareva, N.; Subramanian, D.; Griffith, J.D. Analysis of the binding of p53 to DNAs containing mismatched and bulged bases. J. Biol. Chem. 2001, 276, 8778–8784. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Deppert, W. The complex interactions of p53 with target DNA: We learn as we go. Biochem. Cell Biol. 2003, 81, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Stros, M.; Muselikova-Polanska, E.; Pospisilova, S.; Strauss, F. High-affinity binding of tumor-suppressor protein p53 and HMGB1 to hemicatenated DNA loops. Biochemistry 2004, 43, 7215–7225. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, D.; Griffith, J.D. p53 Monitors replication fork regression by binding to “chickenfoot” intermediates. J. Biol. Chem. 2005, 280, 42568–42572. [Google Scholar] [CrossRef] [PubMed]
- Stansel, R.M.; Subramanian, D.; Griffith, J.D. p53 binds telomeric single strand overhangs and T-loop junctions in vitro. J. Biol. Chem. 2002, 277, 11625–11628. [Google Scholar] [CrossRef] [PubMed]
- Brazda, V.; Palecek, J.; Pospisilova, S.; Vojtesek, B.; Palecek, E. Specific modulation of p53 binding to consensus sequence within supercoiled DNA by monoclonal antibodies. Biochem. Biophys. Res. Commun. 2000, 267, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Palecek, E.; Brazdova, M.; Brazda, V.; Palecek, J.; Billova, S.; Subramaniam, V.; Jovin, T.M. Binding of p53 and its core domain to supercoiled DNA. Eur. J. Biochem. 2001, 268, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Elenbaas, B.; Levine, A.; Griffith, J. p53 and Its 14 kDa C-terminal domain recognize primary DNA-damage in the form of insertion deletion mismatches. Cell 1995, 81, 1013–1020. [Google Scholar] [CrossRef]
- Cobb, A.M.; Jackson, B.R.; Kim, E.; Bond, P.L.; Bowater, R.P. Sequence-specific and DNA structure-dependent interactions of Escherichia coli MutS and human p53 with DNA. Anal. Biochem. 2013, 442, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Cavallo, L.; Griffith, J. Human p53 binds Holliday junctions strongly and facilitates their cleavage. J. Biol. Chem. 1997, 272, 7532–7539. [Google Scholar] [CrossRef] [PubMed]
- Picksley, S.M.; Meek, D.W.; Lane, D.P. The conformational change of a murine temperature-sensitive p53 protein is independent of a change in phosphorylation status. Oncogene 1992, 7, 1649–1651. [Google Scholar] [PubMed]
- Mazur, S.J.; Sakaguchi, K.; Appella, E.; Wang, X.W.; Harris, C.C.; Bohr, V.A. Preferential binding of tumor suppressor p53 to positively or negatively supercoiled DNA involves the C-terminal domain. J. Mol. Biol. 1999, 292, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Pivonkova, H.; Sebest, P.; Pecinka, P.; Ticha, O.; Nemcova, K.; Brazdova, M.; Jagelska, E.B.; Brazda, V.; Fojta, M. Selective binding of tumor suppressor p53 protein to topologically constrained DNA: Modulation by intercalative drugs. Biochem. Biophys. Res. Commun. 2010, 393, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Jett, S.D.; Cherny, D.I.; Subramaniam, V.; Jovin, T.M. Scanning force microscopy of the complexes of p53 core domain with supercoiled DNA. J. Mol. Biol. 2000, 299, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Cherny, D.I.; Striker, G.; Subramaniam, V.; Jett, S.D.; Palecek, E.; Jovin, T.M. DNA bending due to specific p53 and p53 core domain-DNA interactions visualized by electron microscopy. J. Mol. Biol. 1999, 294, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Saramaki, A.; Banwell, C.M.; Campbell, M.J.; Carlberg, C. Regulation of the human p21(waf1/cip1) gene promoter via multiple binding sites for p53 and the vitamin D3 receptor. Nucleic Acids Res. 2006, 34, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, C.; Strauss, F. High affinity binding of proteins HMG1 and HMG2 to semicatenated DNA loops. BMC Mol. Biol. 2000, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; No, Y.R.; Dang, D.T.; Dang, L.H.; Yang, V.W.; Shim, H.; Yun, C.C. Regulation of hypoxia-iinducible factor 1α (HIF-1α) by lysophosphatidic acid is dependent on interplay between p53 and Kruppel-like factor 5. J. Biol. Chem. 2013, 288, 25244–25253. [Google Scholar] [CrossRef] [PubMed]
- Kasparkova, J.; Pospisilova, S.; Brabec, V. Different recognition of DNA modified by aatitumor cisplatin and its clinically ineffective trans isomer by tumor suppressor protein p53. J. Biol. Chem. 2001, 276, 16064–16069. [Google Scholar] [CrossRef] [PubMed]
- Bakalkin, G.; Yakovleva, T.; Selivanova, G.; Magnusson, K.P.; Szekely, L.; Kiseleva, E.; Klein, G.; Terenius, L.; Wiman, K.G. p53 binds single-stranded DNA ends and catalyzes DNA renaturation and strand transfer. Proc. Natl. Acad. Sci. USA 1994, 91, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.J.; Faaland, C.A.; Gallo, M.A.; Thomas, T. Suppression of c-myc oncogene expression by a polyamine-complexed triplex forming oligonucleotide in MCF-7 breast cancer cells. Nucleic Acids Res. 1995, 23, 3594–3599. [Google Scholar] [CrossRef] [PubMed]
- Brazdova, M.; Tichy, V.; Helma, R.; Bazantova, P.; Polaskova, A.; Krejci, A.; Petr, M.; Navratilova, L.; Ticha, O.; Nejedly, K.; et al. p53 specifically binds triplex DNA in vitro and in cells. PLoS ONE 2016, 11, e0167439. [Google Scholar]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [PubMed]
- Haronikova, L.; Coufal, J.; Kejnovska, I.; Jagelska, E.B.; Fojta, M.; Dvorakova, P.; Muller, P.; Vojtesek, B.; Brazda, V. IFI16 preferentially binds to DNA with quadruplex structure and enhances DNA quadruplex formation. PLoS ONE 2016, 11, e0157156. [Google Scholar] [CrossRef] [PubMed]
- Gohler, T.; Jager, S.; Warnecke, G.; Yasuda, H.; Kim, E.; Deppert, W. Mutant p53 proteins bind DNA in a DNA structure-selective mode. Nucleic Acids Res. 2005, 33, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Brazda, V.; Muller, P.; Brozkova, K.; Vojtesek, B. Restoring wild-type conformation and DNA-binding activity of mutant p53 is insufficient for restoration of transcriptional activity. Biochem. Biophys. Res. Commun. 2006, 351, 499–506. [Google Scholar] [CrossRef]
- Quante, T.; Otto, B.; Brazdova, M.; Kejnovska, I.; Deppert, W.; Tolstonog, G.V. Mutant p53 is a transcriptional co-factor that binds to G-rich regulatory regions of active genes and generates transcriptional plasticity. Cell Cycle 2012, 11, 3290–3303. [Google Scholar] [CrossRef] [PubMed]
- Brazdova, M.; Quante, T.; Togel, L.; Walter, K.; Loscher, C.; Tichy, V.; Cincarova, L.; Deppert, W.; Tolstonog, G.V. Modulation of gene expression in U251 glioblastoma cells by binding of mutant p53 R273H to intronic and intergenic sequences. Nucleic Acids Res. 2009, 37, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Chicas, A.; Molina, P.; Bargonetti, J. Mutant p53 forms a complex with Sp1 on HIV-LTR DNA. Biochem. Biophys. Res. Commun. 2000, 279, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Sampath, J.; Sun, D.X.; Kidd, V.J.; Grenet, J.; Gandhi, A.; Shapiro, L.H.; Wang, Q.J.; Zambetti, G.P.; Schuetz, J.D. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J. Biol. Chem. 2001, 276, 39359–39367. [Google Scholar] [CrossRef] [PubMed]
- Petr, M.; Helma, R.; Polaskova, A.; Krejci, A.; Dvorakova, Z.; Kejnovska, I.; Navratilova, L.; Adamik, M.; Vorlickova, M.; Brazdova, M. Wild-type p53 binds to MYC promoter G-quadruplex. Biosci. Rep. 2016, 36, e00397. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.; Warnecke, G.; Bowater, R.; Deppert, W.; Kim, E. Tumor suppressor p53 binds with high affinity to CTG·CAG trinucleotide repeats and induces topological alterations in mismatched duplexes. J. Biol. Chem. 2005, 280, 42497–42507. [Google Scholar] [CrossRef] [PubMed]
- Kamada, R.; Toguchi, Y.; Nomura, T.; Imagawa, T.; Sakaguchi, K. Tetramer formation of tumor suppressor protein p53: Structure, function, and applications. Biopolymers 2016, 106, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Chene, P. The role of tetramerization in p53 function. Oncogene 2001, 20, 2611–2617. [Google Scholar] [CrossRef] [PubMed]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Dell’Orso, S.; Mongiovi, A.M.; Monti, O.; Lapi, E.; Di Agostino, S.; Fontemaggi, G.; Blandino, G. Mutant p53 proteins: Between loss and gain of function. Head Neck 2007, 29, 488–496. [Google Scholar] [CrossRef]
- Noy, A.; Sutthibutpong, T.; Harris, S.A. Protein/DNA interactions in complex DNA topologies: Expect the unexpected. Biophys. Rev. 2016, 8, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Brazda, V.; Haronikova, L.; Liao, J.C.; Fojta, M. DNA and RNA quadruplex-binding proteins. Int. J. Mol. Sci. 2014, 15, 17493–17517. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Deppert, W. The versatile interactions of p53 with DNA: When flexibility serves specificity. Cell Death Differ. 2006, 13, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Deppert, W. Binding of MAR-DNA elements by mutant p53: Possible implications for its oncogenic functions. J. Cell. Biochem. 1996, 62, 172–180. [Google Scholar] [CrossRef]
- Will, K.; Warnecke, G.; Wiesmuller, L.; Deppert, W. Specific interaction of mutant p53 with regions of matrix attachment region DNA elements (MARs) with a high potential for base-unpairing. Proc. Natl. Acad. Sci. USA 1998, 95, 13681–13686. [Google Scholar] [CrossRef] [PubMed]
- Laptenko, O.; Tong, D.R.; Manfredi, J.; Prives, C. The tail that wags the dog: How the disordered C-terminal domain controls the transcriptional activities of the p53 tumor-suppressor protein. Trends Biochem. Sci. 2016, 41, 1022–1034. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Deppert, W. Transcriptional activities of mutant p53: When mutations are more than a loss. J. Cell. Biochem. 2004, 93, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Tutton, S.; Azzam, G.A.; Stong, N.; Vladimirova, O.; Wiedmer, A.; Monteith, J.A.; Beishline, K.; Wang, Z.; Deng, Z.; Riethman, H.; et al. Subtelomeric p53 binding prevents accumulation of DNA damage at human telomeres. EMBO J. 2016, 35, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Kenzelmann Broz, D.; Spano Mello, S.; Bieging, K.T.; Jiang, D.; Dusek, R.L.; Brady, C.A.; Sidow, A.; Attardi, L.D. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013, 27, 1016–1031. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brázda, V.; Coufal, J. Recognition of Local DNA Structures by p53 Protein. Int. J. Mol. Sci. 2017, 18, 375. https://doi.org/10.3390/ijms18020375
Brázda V, Coufal J. Recognition of Local DNA Structures by p53 Protein. International Journal of Molecular Sciences. 2017; 18(2):375. https://doi.org/10.3390/ijms18020375
Chicago/Turabian StyleBrázda, Václav, and Jan Coufal. 2017. "Recognition of Local DNA Structures by p53 Protein" International Journal of Molecular Sciences 18, no. 2: 375. https://doi.org/10.3390/ijms18020375
APA StyleBrázda, V., & Coufal, J. (2017). Recognition of Local DNA Structures by p53 Protein. International Journal of Molecular Sciences, 18(2), 375. https://doi.org/10.3390/ijms18020375