
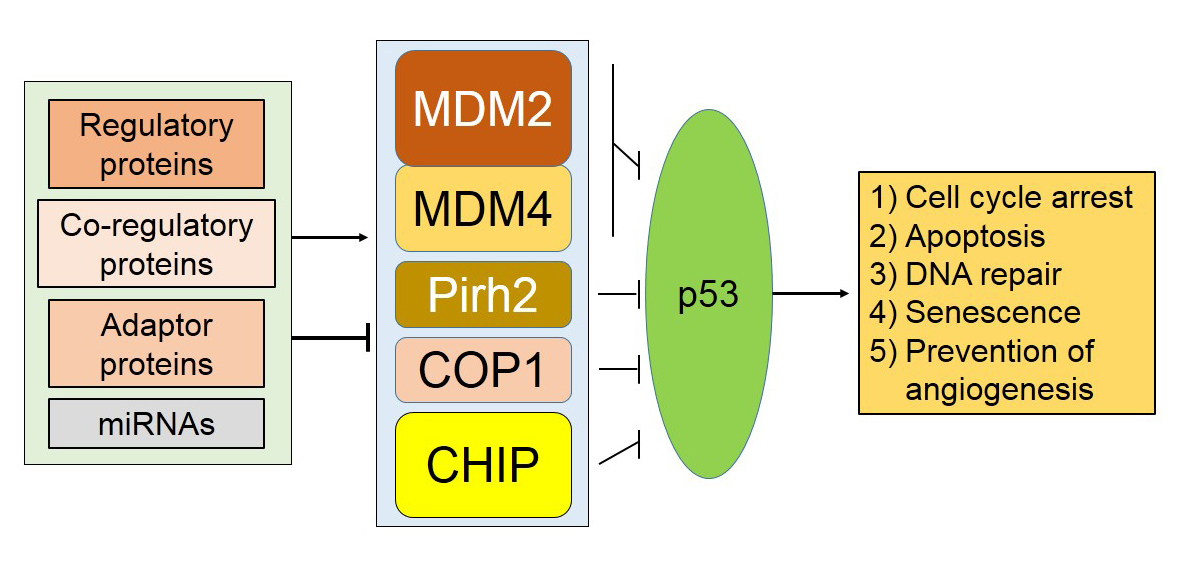
Essential Roles of E3 Ubiquitin Ligases in p53 Regulation

Abstract
:
1. Introduction
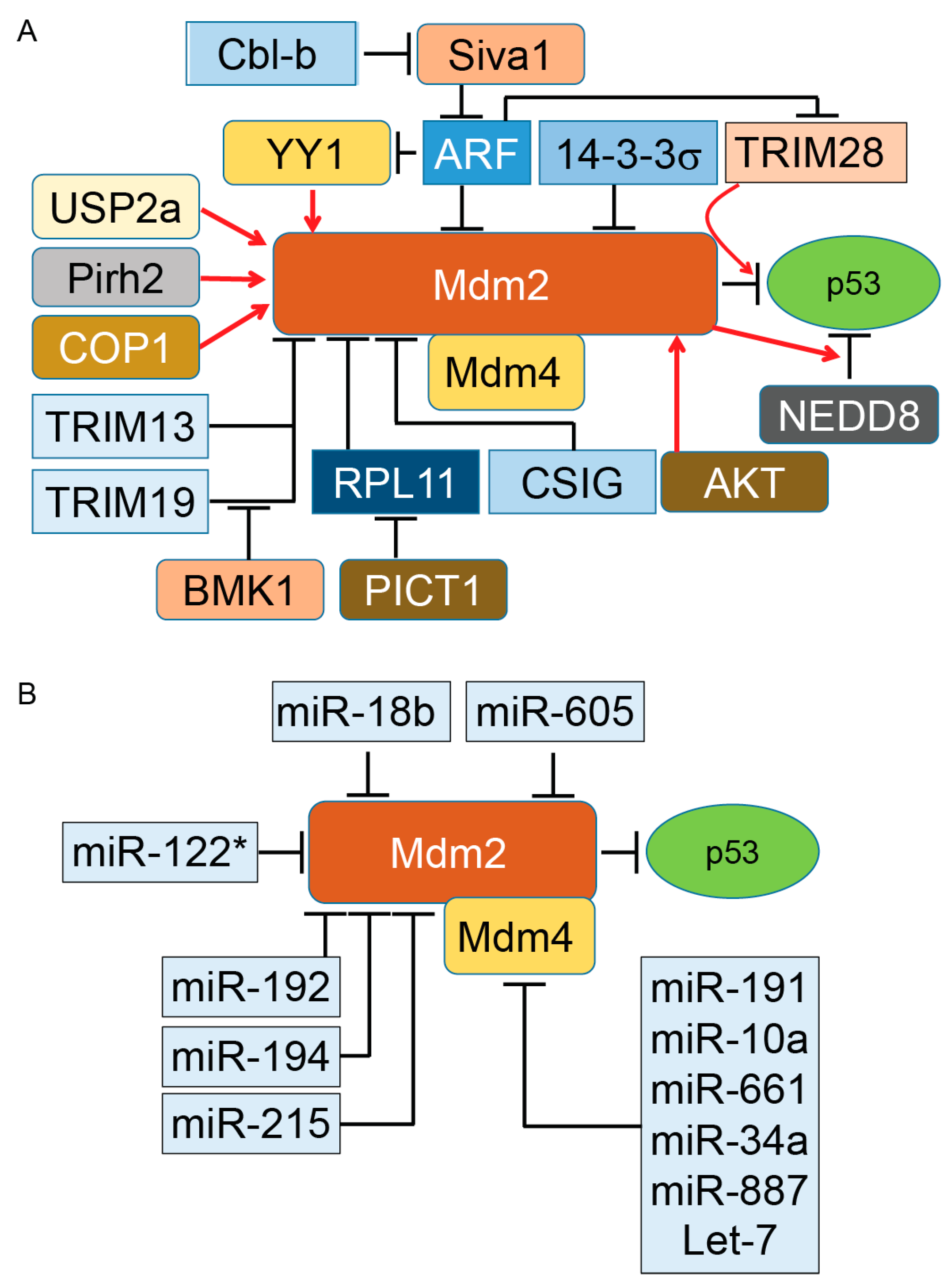
2. Murine Double Minute (Mdm)2/Mdm4, a Dominant E3 Ligase of p53
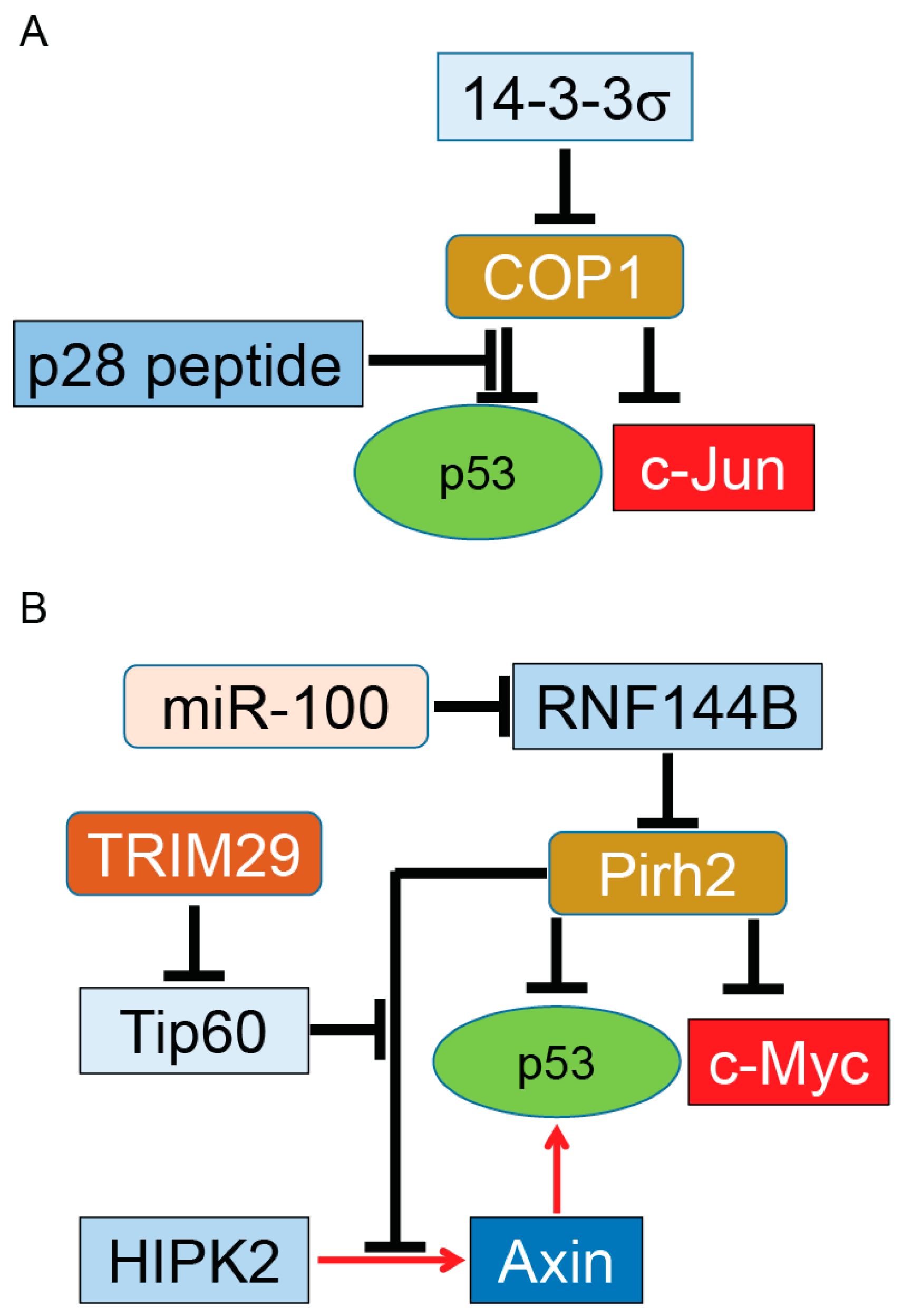
3. Constitutive Photomorphogenesis Protein 1 (COP1) and P53 Regulation
4. Pirh2 (p53-Induced Protein with a RING-H2 Domain) and p53 Regulation
5. Co-Chaperone Carboxyl Terminus Hsp70/90 Interacting Protein (CHIP) E3 Ubiquitin Ligase
6. Discussion
Acknowledgments
Conflicts of Interest
References
- Jenkins, L.M.; Durell, S.R.; Mazur, S.J.; Appella, E. p53 N-terminal phosphorylation: A defining layer of complex regulation. Carcinogenesis 2012, 33, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.L.; Veprintsev, D.B.; Bycroft, M.; Fersht, A.R. Comparative binding of p53 to its promoter and DNA recognition elements. J. Mol. Biol. 2005, 348, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef] [PubMed]
- Chene, P. The role of tetramerization in p53 function. Oncogene 2001, 20, 2611–2617. [Google Scholar] [CrossRef] [PubMed]
- Bakalkin, G.; Selivanova, G.; Yakovleva, T.; Kiseleva, E.; Kashuba, E.; Magnusson, K.P.; Szekely, L.; Klein, G.; Terenius, L.; Wiman, K.G. p53 binds single-stranded DNA ends through the C-terminal domain and internal DNA segments via the middle domain. Nucleic Acids Res. 1995, 23, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- O’Brate, A.; Giannakakou, P. The importance of p53 location: Nuclear or cytoplasmic zip code? Drug Resist. Updates 2003, 6, 313–322. [Google Scholar] [CrossRef]
- Ashcroft, M.; Taya, Y.; Vousden, K.H. Stress signals utilize multiple pathways to stabilize p53. Mol. Cell. Biol. 2000, 20, 3224–3233. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Wu, L.; Stuart, J.; Maki, C.G. Control of p53 nuclear accumulation in stressed cells. FEBS Lett. 2005, 579, 4978–4984. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Maki, C.G. p53 Localization. In p53; Springer: Boston, MA, USA, 2010; pp. 117–126. [Google Scholar]
- Lane, D.P.; Cheok, C.F.; Lain, S. p53-based cancer therapy. Cold Spring Harb. Perspect. Biol. 2010, 2, a001222. [Google Scholar] [CrossRef] [PubMed]
- Bossi, G.; Sacchi, A. Restoration of wild-type p53 function in human cancer: Relevance for tumor therapy. Head Neck 2007, 29, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zeng, S.X.; Lu, H. Targeting p53–Mdm2–Mdmx loop for cancer therapy. Subcell. Biochem. 2014, 85, 281–319. [Google Scholar] [PubMed]
- Freedman, D.A.; Wu, L.; Levine, A.J. Functions of the Mdm2 oncoprotein. Cell. Mol. Life Sci. 1999, 55, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Momand, J.; Wu, H.H.; Dasgupta, G. Mdm2—Master regulator of the p53 tumor suppressor protein. Gene 2000, 242, 15–29. [Google Scholar] [CrossRef]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53–Mdm2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono-versus polyubiquitination: Differential control of p53 fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Li, Y.C.; Matani, A.S.; Braun, S.M.; Milanesi, F.; Rodewald, L.W.; Wahl, G.M. Functional analysis and consequences of Mdm2 E3 ligase inhibition in human tumor cells. Oncogene 2012, 31, 4789–4797. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Sasaki, M.; Maki, C.G. Regulation of p53 nuclear export through sequential changes in conformation and ubiquitination. J. Biol. Chem. 2007, 282, 14616–14625. [Google Scholar] [CrossRef] [PubMed]
- Geyer, R.K.; Yu, Z.K.; Maki, C.G. The Mdm2 RING-finger domain is required to promote p53 nuclear export. Nat. Cell Biol. 2000, 2, 569–573. [Google Scholar] [PubMed]
- Jones, S.N.; Hancock, A.R.; Vogel, H.; Donehower, L.A.; Bradley, A. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 15608–15612. [Google Scholar] [CrossRef] [PubMed]
- Bouska, A.; Eischen, C.M. Murine double minute 2: p53-independent roads lead to genome instability or death. Trends Biochem. Sci. 2009, 34, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Toguchida, J.; Wadayama, B.; Kanoe, H.; Kotoura, Y.; Sasaki, M.S. Mdm2 gene amplification in bone and soft-tissue tumors: Association with tumor progression in differentiated adipose-tissue tumors. Int. J. Cancer 1995, 64, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Cordon-Cardo, C.; Latres, E.; Drobnjak, M.; Oliva, M.R.; Pollack, D.; Woodruff, J.M.; Marechal, V.; Chen, J.; Brennan, M.F.; Levine, A.J. Molecular abnormalities of Mdm2 and p53 genes in adult soft tissue sarcomas. Cancer Res. 1994, 54, 794–799. [Google Scholar] [PubMed]
- Vassilev, L.T. Mdm2 inhibitors for cancer therapy. Trends Mol. Med. 2007, 13, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.W.; Aslo, A.; Won, A.; Tan, M.; Lampkin, B.; Koeffler, H.P. Alterations of the p53, Rb and Mdm2 genes in osteosarcoma. J. Cancer Res. Clin. Oncol. 1996, 122, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, G.; Liu, L.; Ichimura, K.; Schmidt, E.E.; Collins, V.P. Amplification and overexpression of the Mdm2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res. 1993, 53, 2736–2739. [Google Scholar] [PubMed]
- Watanabe, T.; Hotta, T.; Ichikawa, A.; Kinoshita, T.; Nagai, H.; Uchida, T.; Murate, T.; Saito, H. The Mdm2 oncogene overexpression in chronic lymphocytic leukemia and low-grade lymphoma of B-cell origin. Blood 1994, 84, 3158–3165. [Google Scholar] [PubMed]
- Rayburn, E.; Zhang, R.; He, J.; Wang, H. Mdm2 and human malignancies: Expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr. Cancer Drug Targets 2005, 5, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical overview of Mdm2/x-targeted therapies. Front. Oncol. 2016, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Konopleva, M.; Samudio, I.J.; Shikami, M.; Cabreira-Hansen, M.; McQueen, T.; Ruvolo, V.; Tsao, T.; Zeng, Z.; Vassilev, L.T.; et al. Mdm2 antagonists induce p53-dependent apoptosis in AML: Implications for leukemia therapy. Blood 2005, 106, 3150–3159. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; di Iasio, M.G.; Gonelli, A.; Zauli, G. The Mdm2 inhibitor Nutlins as an innovative therapeutic tool for the treatment of haematological malignancies. Curr. Pharm. Des. 2008, 14, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Van Maerken, T.; Ferdinande, L.; Taildeman, J.; Lambertz, I.; Yigit, N.; Vercruysse, L.; Rihani, A.; Michaelis, M.; Cinatl, J., Jr.; Cuvelier, C.A.; et al. Antitumor activity of the selective Mdm2 antagonist Nutlin-3 against chemoresistant neuroblastoma with wild-type p53. J. Natl. Cancer Inst. 2009, 101, 1562–1574. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Bosco, R.; Celeghini, C.; Zauli, G. Recent advances in the therapeutic perspectives of Nutlin-3. Curr. Pharm. Des. 2011, 17, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yu, H.; Hu, W. The regulation of Mdm2 oncogene and its impact on human cancers. Acta Biochim. Biophys. Sin. 2014, 46, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Cheok, C.F.; Verma, C.S.; Baselga, J.; Lane, D.P. Translating p53 into the clinic. Nat. Rev. Clin. Oncol. 2011, 8, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.N.; Roe, A.E.; Donehower, L.A.; Bradley, A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995, 378, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Bunderson-Schelvan, M.; Erbe, A.K.; Schwanke, C.; Pershouse, M.A. Suppression of the mouse double minute 4 gene causes changes in cell cycle control in a human mesothelial cell line responsive to ultraviolet radiation exposure. Environ. Mol. Mutagen. 2009, 50, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Wang, Y.V.; Wahl, G.M. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 2010, 20, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, J.; Jiang, X. MdmX protein is essential for Mdm2 protein-mediated p53 polyubiquitination. J. Biol. Chem. 2011, 286, 23725–23734. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lozano, G. Molecular pathways: Targeting Mdm2 and Mdm4 in cancer therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, G.; Zhang, P.; Wang, X.; Jiang, M.; Yu, L. Interplay between Mdm2, Mdmx, Pirh2 and COP1: The negative regulators of p53. Mol. Biol. Rep. 2011, 38, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. ARF promotes Mdm2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998, 92, 725–734. [Google Scholar] [CrossRef]
- Sherr, C.J.; Weber, J.D. The ARF/p53 pathway. Curr. Opin. Genet. Dev. 2000, 10, 94–99. [Google Scholar] [CrossRef]
- Christophorou, M.A.; Ringshausen, I.; Finch, A.J.; Swigart, L.B.; Evan, G.I. The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature 2006, 443, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Murga, M.; Martinez-Pastor, B.; Ortega-Molina, A.; Soria, R.; Collado, M.; Fernandez-Capetillo, O.; Serrano, M. Limited role of murine ATM in oncogene-induced senescence and p53-dependent tumor suppression. PLoS ONE 2009, 4, e5475. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zha, M.; Zhao, X.; Jiang, P.; Du, W.; Tam, A.Y.; Mei, Y.; Wu, M. Siva1 inhibits p53 function by acting as an ARF E3 ubiquitin ligase. Nat. Commun. 2013, 4, 1551. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Blum, W.; Baker, S.D.; Caligiuri, M.A. E3 ubiquitin ligase Cbl-b activates the p53 pathway by targeting Siva1, a negative regulator of ARF, in FLT3 inhibitor-resistant acute myeloid leukemia. Leukemia 2016, 31, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ivanov, A.; Chen, L.; Fredericks, W.J.; Seto, E.; Rauscher, F.J.; Chen, J. Mdm2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J. 2005, 24, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.M.; Kim, J.Y.; Jeong, J.B.; Seong, K.M.; Nam, S.Y.; Yang, K.H.; Kim, C.S.; Kim, H.S.; Jeong, M.; An, S.; et al. Ret finger protein 2 enhances ionizing radiation-induced apoptosis via degradation of AKT and Mdm2. Eur. J. Cell Biol. 2011, 90, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.; Scaglioni, P.P.; Bergmann, S.; Horn, H.F.; Vousden, K.H.; Pandolfi, P.P. PML regulates p53 stability by sequestering Mdm2 to the nucleolus. Nat. Cell Biol. 2004, 6, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Ma, L.; Zhu, F.; Zhao, W.; Tian, F.; Yuan, F.; Fu, J.; Huang, D.; Lv, C.; Tong, T. Regulation of the Mdm2-p53 pathway by the nucleolar protein CSIG in response to nucleolar stress. Sci. Rep. 2016, 6, 36171. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liao, L.; Deng, X.; Chen, R.; Gray, N.S.; Yates, J.R.; Lee, J.D. BMK1 is involved in the regulation of p53 through disrupting the PML-Mdm2 interaction. Oncogene 2013, 32, 3156–3164. [Google Scholar] [CrossRef] [PubMed]
- Simerzin, A.; Zorde-Khvalevsky, E.; Rivkin, M.; Adar, R.; Zucman-Rossi, J.; Couchy, G.; Roskams, T.; Govaere, O.; Oren, M.; Giladi, H.; et al. The liver-specific microRNA-122*, the complementary strand of microRNA-122, acts as a tumor suppressor by modulating the p53/mouse double minute 2 homolog circuitry. Hepatology 2016, 64, 1623–1636. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R.S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; et al. Temporal activation of p53 by a specific Mdm2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 3933–3938. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Konopleva, M.; McQueen, T.; O’Brien, S.; Plunkett, W.; Andreeff, M. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome ATM-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood 2006, 108, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Pichiorri, F.; Suh, S.-S.; Rocci, A.; De Luca, L.; Taccioli, C.; Santhanam, R.; Zhou, W.; Benson, D.M., Jr.; Hofmainster, C.; Alder, H.; et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 Impairs the p53/Mdm2 Autoregulatory Loop in Multiple Myeloma Development. Cancer Cell 2010, 18, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Lin, H.; Luo, X.; Luo, X.; Wang, Z. miR-605 joins p53 network to form a p53:miR-605:Mdm2 positive feedback loop in response to stress. EMBO J. 2011, 30, 5021. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.A.; Majid, S.; Rittsteuer, C.; de Semir, D.; Bezrookove, V.; Tong, S.; Nosrati, M.; Sagebiel, R.; Miller, J.R., III; Kashani-Sabet, M. The role of miR-18b in Mdm2-p53 pathway signaling and melanoma progression. J. Natl. Cancer Inst. 2013, 105, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Wynendaele, J.; Bohnke, A.; Leucci, E.; Nielsen, S.J.; Lambertz, I.; Hammer, S.; Sbrzesny, N.; Kubitza, D.; Wolf, A.; Gradhand, E.; et al. An illegitimate microRNA target site within the 3′ UTR of Mdm4 affects ovarian cancer progression and chemosensitivity. Cancer Res. 2010, 70, 9641–9649. [Google Scholar] [CrossRef] [PubMed]
- Ovcharenko, D.; Stolzel, F.; Poitz, D.; Fierro, F.; Schaich, M.; Neubauer, A.; Kelnar, K.; Davison, T.; Muller-Tidow, C.; Thiede, C.; et al. miR-10a overexpression is associated with NPM1 mutations and Mdm4 downregulation in intermediate-risk acute myeloid leukemia. Exp. Hematol. 2011, 39, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, Y.; Bublik, D.R.; Pilpel, Y.; Oren, M. miR-661 downregulates both Mdm2 and Mdm4 to activate p53. Cell Death Differ. 2014, 21, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Mandke, P.; Wyatt, N.; Fraser, J.; Bates, B.; Berberich, S.J.; Markey, M.P. MicroRNA-34a modulates Mdm4 expression via a target site in the open reading frame. PLoS ONE 2012, 7, e42034. [Google Scholar] [CrossRef] [PubMed]
- Stegeman, S.; Moya, L.; Selth, L.A.; Spurdle, A.B.; Clements, J.A.; Batra, J. A genetic variant of Mdm4 influences regulation by multiple microRNAs in prostate cancer. Endocr. Relat. Cancer 2015, 22, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Chen, W.; Zhang, M.; Cai, Q.; Xu, W.; Li, X.; Jiang, S. Mdm4 regulation by the let-7 miRNA family in the DNA damage response of glioma cells. FEBS Lett. 2015, 589, 1958–1965. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumaran, R.; Tan, K.H.; Miranda, P.J.; Haupt, S.; Haupt, Y. Regulation of mutant p53 protein expression. Front. Oncol. 2015, 5, 284. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, Y.; Pilpel, Y.; Oren, M. microRNAs and Alu elements in the p53–Mdm2–Mdm4 regulatory network. J. Mol. Cell Biol. 2014, 6, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.Y.; Wen, Y.Y.; Chen, C.H.; Lozano, G.; Lee, M.H. 14-3-3σ positively regulates p53 and suppresses tumor growth. Mol. Cell. Biol. 2003, 23, 7096–7107. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, L.F.; Sparks, A.; Allende-Vega, N.; Xirodimas, D.P.; Lane, D.P.; Saville, M.K. The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO J. 2007, 26, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wolf, G.W.; Bhat, K.; Jin, A.; Allio, T.; Burkhart, W.A.; Xiong, Y. Ribosomal protein L11 negatively regulates oncoprotein Mdm2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell. Biol. 2003, 23, 8902–8912. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Lang, Y.; Zhang, Q.; Cui, D.; Sun, H.; Jiang, L.; Chen, Z.; Zhang, R.; Gao, Y.; Tian, W.; et al. Structure of human Mdm2 complexed with RPL11 reveals the molecular basis of p53 activation. Genes Dev. 2015, 29, 1524–1534. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Kawahara, K.; Nishio, M.; Mimori, K.; Kogo, R.; Hamada, K.; Itoh, B.; Wang, J.; Komatsu, Y.; Yang, Y.R.; et al. Regulation of the Mdm2–P53 pathway and tumor growth by PICT1 via nucleolar RPL11. Nat. Med. 2011, 17, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Sui, G.; Affar el, B.; Shi, Y.; Brignone, C.; Wall, N.R.; Yin, P.; Donohoe, M.; Luke, M.P.; Calvo, D.; Grossman, S.R.; et al. Yin Yang 1 is a negative regulator of p53. Cell 2004, 117, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, T.M.; Leal, J.F.; Seger, R.; Taya, Y.; Oren, M. Cross-talk between Akt, p53 and Mdm2: Possible implications for the regulation of apoptosis. Oncogene 2002, 21, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Xirodimas, D.P.; Saville, M.K.; Bourdon, J.C.; Hay, R.T.; Lane, D.P. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell 2004, 118, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Koegl, M.; Hoppe, T.; Schlenker, S.; Ulrich, H.D.; Mayer, T.U.; Jentsch, S. A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 1999, 96, 635–644. [Google Scholar] [CrossRef]
- Shi, D.; Pop, M.S.; Kulikov, R.; Love, I.M.; Kung, A.L.; Grossman, S.R. CBP and p300 are cytoplasmic E4 polyubiquitin ligases for p53. Proc. Natl. Acad. Sci. USA 2009, 106, 16275–16280. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.R.; Deato, M.E.; Brignone, C.; Chan, H.M.; Kung, A.L.; Tagami, H.; Nakatani, Y.; Livingston, D.M. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science 2003, 300, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Pomeroy, S.L.; Ferreira, M.; Teider, N.; Mariani, J.; Nakayama, K.I.; Hatakeyama, S.; Tron, V.A.; Saltibus, L.F.; Spyracopoulos, L.; et al. UBE4B promotes Hdm2-mediated degradation of the tumor suppressor p53. Nat. Med. 2011, 17, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.W.; Caspar, T.; Quail, P.H. cop1: A regulatory locus involved in light-controlled development and gene expression in Arabidopsis. Genes Dev. 1991, 5, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, E.; Denti, S.; Catena, R.; Rossetti, G.; Polo, S.; Gasparian, S.; Putignano, S.; Rogge, L.; Pardi, R. Characterization of human constitutive photomorphogenesis protein 1, a RING finger ubiquitin ligase that interacts with Jun transcription factors and modulates their transcriptional activity. J. Biol. Chem. 2003, 278, 19682–19690. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.-C. Spotlight on the role of COP1 in tumorigenesis. Nat. Rev. Cancer 2012, 12, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Wang, D.D.; Zhao, B.W.; Wang, W.; Huang, C.Y.; Chen, Y.M.; Zheng, Y.; Keshari, R.P.; Xia, J.C.; Zhou, Z.W. High level of COP1 expression is associated with poor prognosis in primary gastric cancer. Int. J. Biol. Sci. 2012, 8, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, L.; Xiao, R.; Pan, Q.; Huang, H.; Kuang, R. High expression of constitutive photomorphogenic 1 (COP1) is associated with poor prognosis in bladder cancer. Tumour Biol. 2016, 37, 8917–8922. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Zhu, Y.; Wang, B.; Qian, F.; Zhang, X.; Wang, L.; Fu, C.; Bao, H.; Xie, M.; Gao, S.; et al. The ubiquitin ligase COP1 promotes glioma cell proliferation by preferentially downregulating tumor suppressor p53. Mol. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Dornan, D.; Bheddah, S.; Newton, K.; Ince, W.; Frantz, G.D.; Dowd, P.; Koeppen, H.; Dixit, V.M.; French, D.M. COP1, the negative regulator of p53, is overexpressed in breast and ovarian adenocarcinomas. Cancer Res. 2004, 64, 7226–7230. [Google Scholar] [CrossRef] [PubMed]
- Dornan, D.; Wertz, I.; Shimizu, H.; Arnott, D.; Frantz, G.D.; Dowd, P.; O’Rourke, K.; Koeppen, H.; Dixit, V.M. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 2004, 429, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Vleugel, M.M.; Greijer, A.E.; Bos, R.; van der Wall, E.; van Diest, P.J. c-Jun activation is associated with proliferation and angiogenesis in invasive breast cancer. Hum. Pathol. 2006, 37, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; O’Rourke, K.M.; Zhang, Z.; Dornan, D.; Arnott, D.; Deshaies, R.J.; Dixit, V.M. Human De-etiolated-1 regulates c-Jun by assembling a CUL4A ubiquitin ligase. Science 2004, 303, 1371–1374. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Teng, Y.; Padia, R.; Hong, S.; Noh, H.; Xie, X.; Mumm, J.S.; Dong, Z.; Ding, H.F.; Cowell, J.; et al. COP1 and GSK3β cooperate to promote c-Jun degradation and inhibit breast cancer cell tumorigenesis. Neoplasia 2013, 15, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Andersen, J.B.; Song, H.T.; Judge, A.D.; Seo, D.; Ishikawa, T.; Marquardt, J.U.; Kitade, M.; Durkin, M.E.; Raggi, C.; et al. Definition of ubiquitination modulator COP1 as a novel therapeutic target in human hepatocellular carcinoma. Cancer Res. 2010, 70, 8264–8269. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, D.; Bogaerts, S.; Defever, D.; Vyas, R.; Denecker, G.; Radaelli, E.; Zwolinska, A.; Depaepe, V.; Hochepied, T.; Skarnes, W.C.; et al. COP1 constitutively regulates c-Jun protein stability and functions as a tumor suppressor in mice. J. Clin. Investig. 2011, 121, 1329–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.H.; Zhao, R.; Zhang, F.; Qu, C.; Chen, B.; Feng, Y.H.; Phan, L.; Chen, J.; Wang, H.; Wang, H.; et al. 14-3-3σ exerts tumor-suppressor activity mediated by regulation of COP1 stability. Cancer Res. 2011, 71, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Gorman, G.S.; Coward, L.U.; Noker, P.E.; McCormick, D.; Horn, T.L.; Harder, J.B.; Muzzio, M.; Prabhakar, B.; Ganesh, B.; et al. Preclinical pharmacokinetics, metabolism, and toxicity of azurin-p28 (NSC745104) a peptide inhibitor of p53 ubiquitination. Cancer Chemother. Pharmacol. 2011, 68, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Apiyo, D.; Wittung-Stafshede, P. Unique complex between bacterial azurin and tumor-suppressor protein p53. Biochem. Biophys. Res. Commun. 2005, 332, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Christov, K.; Shilkaitis, A.; Bratescu, L.; Green, A.; Santini, S.; Bizzarri, A.R.; Cannistraro, S.; Gupta, T.K.; Beattie, C.W. p28, a first in class peptide inhibitor of cop1 binding to p53. Br. J. Cancer 2013, 108, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Leng, R.P.; Lin, Y.; Ma, W.; Wu, H.; Lemmers, B.; Chung, S.; Parant, J.M.; Lozano, G.; Hakem, R.; Benchimol, S. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 2003, 112, 779–791. [Google Scholar] [CrossRef]
- Chen, J.; Marechal, V.; Levine, A.J. Mapping of the p53 and Mdm-2 interaction domains. Mol. Cell. Biol. 1993, 13, 4107–4114. [Google Scholar] [CrossRef] [PubMed]
- Picksley, S.M.; Vojtesek, B.; Sparks, A.; Lane, D.P. Immunochemical analysis of the interaction of p53 with Mdm2—Fine mapping of the Mdm2 binding site on p53 using synthetic peptides. Oncogene 1994, 9, 2523–2529. [Google Scholar] [PubMed]
- Poyurovsky, M.V.; Katz, C.; Laptenko, O.; Beckerman, R.; Lokshin, M.; Ahn, J.; Byeon, I.J.; Gabizon, R.; Mattia, M.; Zupnick, A.; et al. The C terminus of p53 binds the N-terminal domain of Mdm2. Nat. Struct. Mol. Biol. 2010, 17, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Laister, R.; Lemak, A.; Wu, B.; Tai, E.; Duan, S.; Lukin, J.; Sunnerhagen, M.; Srisailam, S.; Karra, M.; et al. Molecular basis of Pirh2-mediated p53 ubiquitylation. Nat. Struct. Mol. Biol. 2008, 15, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, C.-C.H.; Weissman, A.M. Regulating the p53 system through ubiquitination. Oncogene 2004, 23, 2096–2106. [Google Scholar] [CrossRef] [PubMed]
- Logan, I.R.; Sapountzi, V.; Gaughan, L.; Neal, D.E.; Robson, C.N. Control of human PIRH2 protein stability: Involvement of Tip60 and the proteasome. J. Biol. Chem. 2004, 279, 11696–11704. [Google Scholar] [CrossRef] [PubMed]
- Hakem, A.; Bohgaki, M.; Lemmers, B.; Tai, E.; Salmena, L.; Matysiak-Zablocki, E.; Jung, Y.S.; Karaskova, J.; Kaustov, L.; Duan, S.; et al. Role of Pirh2 in mediating the regulation of p53 and c-Myc. PLoS Genet. 2011, 7, e1002360. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lin, S.; Wang, X.; Lian, G.; Lu, Z.; Guo, H.; Ruan, K.; Wang, Y.; Ye, Z.; Han, J.; et al. Axin determines cell fate by controlling the p53 activation threshold after DNA damage. Nat. Cell Biol. 2009, 11, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Sho, T.; Tsukiyama, T.; Sato, T.; Kondo, T.; Cheng, J.; Saku, T.; Asaka, M.; Hatakeyama, S. TRIM29 negatively regulates p53 via inhibition of Tip60. Biochim. Biophys. Acta 2011, 1813, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Gong, Y.; Wang, Q.; Wang, L.; Zhang, X. miR-100 antagonism triggers apoptosis by inhibiting ubiquitination-mediated p53 degradation. Oncogene 2016. [Google Scholar] [CrossRef] [PubMed]
- Logan, I.R.; Gaughan, L.; McCracken, S.R.; Sapountzi, V.; Leung, H.Y.; Robson, C.N. Human PIRH2 enhances androgen receptor signaling through inhibition of histone deacetylase 1 and is overexpressed in prostate cancer. Mol. Cell. Biol. 2006, 26, 6502–6510. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Gao, L.; Druhan, L.J.; Zhu, W.G.; Morrison, C.; Otterson, G.A.; Villalona-Calero, M.A. Expression of Pirh2, a newly identified ubiquitin protein ligase, in lung cancer. J. Natl. Cancer Inst. 2004, 96, 1718–1721. [Google Scholar] [CrossRef] [PubMed]
- Kossatz, U.; Malek, N.P. p27: Tumor suppressor and oncogene...? Cell Res. 2007, 17, 832–833. [Google Scholar] [CrossRef] [PubMed]
- McDonough, H.; Patterson, C. CHIP: A link between the chaperone and proteasome systems. Cell Stress Chaperones 2003, 8, 303–308. [Google Scholar] [CrossRef]
- Esser, C.; Scheffner, M.; Hohfeld, J. The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J. Biol. Chem. 2005, 280, 27443–27448. [Google Scholar] [CrossRef] [PubMed]
- Elengoe, A.; Naser, M.A.; Hamdan, S. A novel protein interaction between nucleotide binding domain of Hsp70 and p53 motif. Int. J. Genom. 2015, 2015, 391293. [Google Scholar] [CrossRef] [PubMed]
- Fourie, A.M.; Hupp, T.R.; Lane, D.P.; Sang, B.C.; Barbosa, M.S.; Sambrook, J.F.; Gething, M.J.H. Hsp70 binding sites in the tumor suppressor protein p53. J. Biol. Chem. 1997, 272, 19471–19479. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, M.; Nikolay, R.; Rybin, V.; Mayer, M.P. CHIP participates in protein triage decisions by preferentially ubiquitinating Hsp70-bound substrates. FEBS J. 2010, 277, 3353–3367. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.; Hrstka, R.; Coomber, D.; Lane, D.P.; Vojtesek, B. Chaperone-dependent stabilization and degradation of p53 mutants. Oncogene 2008, 27, 3371–3383. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous Mdm2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Davenport, J.; Manjarrez, J.R.; Peterson, L.; Krumm, B.; Blagg, B.S.; Matts, R.L. Gambogic acid, a natural product inhibitor of Hsp90. J. Nat. Prod. 2011, 74, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, Q.; Qi, Q.; Gu, H.Y.; Rong, J.J.; Mu, R.; Zou, M.J.; Tao, L.; You, Q.D.; Guo, Q.L. Gambogic acid-induced degradation of mutant p53 is mediated by proteasome and related to CHIP. J. Cell. Biochem. 2011, 112, 509–519. [Google Scholar] [CrossRef] [PubMed]
- McDonough, H.; Charles, P.C.; Hilliard, E.G.; Qian, S.B.; Min, J.N.; Portbury, A.; Cyr, D.M.; Patterson, C. Stress-dependent Daxx-CHIP interaction suppresses the p53 apoptotic program. J. Biol. Chem. 2009, 284, 20649–20659. [Google Scholar] [CrossRef] [PubMed]
- Sisoula, C.; Trachana, V.; Patterson, C.; Gonos, E.S. CHIP-dependent p53 regulation occurs specifically during cellular senescence. Free Radic. Biol. Med. 2011, 50, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Naito, A.T.; Okada, S.; Minamino, T.; Iwanaga, K.; Liu, M.L.; Sumida, T.; Nomura, S.; Sahara, N.; Mizoroki, T.; Takashima, A.; et al. Promotion of CHIP-mediated p53 degradation protects the heart from ischemic injury. Circ. Res. 2010, 106, 1692–1702. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Rosser, M.F.; Washburn, E.; Muchowski, P.J.; Patterson, C.; Cyr, D.M. Chaperone functions of the E3 ubiquitin ligase CHIP. J. Biol. Chem. 2007, 282, 22267–22277. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Ali, A.; Bhat, R.; Pati, U. CHIP chaperones wild type p53 tumor suppressor protein. J. Biol. Chem. 2007, 282, 28441–28454. [Google Scholar] [CrossRef] [PubMed]
- Black, J.D.; Rezvani, K. Heat shock protein 70s as potential molecular targets for colon cancer therapeutics. Curr. Med. Chem. 2016, 23, 3171–3188. [Google Scholar] [CrossRef] [PubMed]
- Kaul, S.C.; Deocaris, C.C.; Wadhwa, R. Three faces of mortalin: A housekeeper, guardian and killer. Exp. Gerontol. 2007, 42, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, R.; Ryu, J.; Ahn, H.M.; Saxena, N.; Chaudhary, A.; Yun, C.O.; Kaul, S.C. Functional significance of point mutations in stress chaperone mortalin and their relevance to Parkinson disease. J. Biol. Chem. 2015, 290, 8447–8456. [Google Scholar] [CrossRef] [PubMed]
- Sane, S.; Abdullah, A.; Boudreau, D.A.; Autenried, R.K.; Gupta, B.K.; Wang, X.; Wang, H.; Schlenker, E.H.; Zhang, D.; Telleria, C.; et al. Ubiquitin-like (UBX)-domain-containing protein, UBXN2A, promotes cell death by interfering with the p53-Mortalin interactions in colon cancer cells. Cell Death Dis. 2014, 5, e1118. [Google Scholar] [CrossRef] [PubMed]
- Sane, S.; Abdullah, A.; Nelson, M.E.; Wang, H.; Chauhan, S.C.; Newton, S.S.; Rezvani, K. Structural studies of UBXN2A and mortalin interaction and the putative role of silenced UBXN2A in preventing response to chemotherapy. Cell Stress Chaperones 2016, 21, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Ryan, K.M. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Peller, S.; Rotter, V. TP53 in hematological cancer: Low incidence of mutations with significant clinical relevance. Hum. Mutat. 2003, 21, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Schuijer, M.; Berns, E.M. TP53 and ovarian cancer. Hum. Mutat. 2003, 21, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacopetta, B. TP53 mutation in colorectal cancer. Hum. Mutat. 2003, 21, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Lain, S.; Lane, D. Improving cancer therapy by non-genotoxic activation of p53. Eur. J. Cancer 2003, 39, 1053–1060. [Google Scholar] [CrossRef]
- Jain, A.K.; Barton, M.C. Making sense of ubiquitin ligases that regulate p53. Cancer Biol. Ther. 2010, 10, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. p53 regulation by ubiquitin. FEBS Lett. 2011, 585, 2803–2809. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Gu, W. The multiple levels of regulation by p53 ubiquitination. Cell Death Differ. 2010, 17, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Mackay, H.; Hedley, D.; Major, P.; Townsley, C.; Mackenzie, M.; Vincent, M.; Degendorfer, P.; Tsao, M.S.; Nicklee, T.; Birle, D.; et al. A phase II trial with pharmacodynamic endpoints of the proteasome inhibitor bortezomib in patients with metastatic colorectal cancer. Clin. Cancer Res. 2005, 11, 5526–5533. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UniProt ID | Protein Symbol | Protein Names | Length (aa) | Binding Site on p53 | Class of E3 Ubiquitin Ligase |
|---|---|---|---|---|---|
| Q00987 | Mdm2 | Mouse double minute 2 homolog (Mdm2) | 491 | 1–51 aa and C-terminus | RING-finger containing E3 ubiqutin ligase |
| Q8NHY2 | COP1 | Constitutively photomorphogenic 1 | 731 | Regions within the DNA-binding domain | RING-finger containing E3 ubiqutin ligase |
| Q96PM5 | Pirh2 | p53-induced RING-H2 protein | 261 | 82–292 aa and the tetramerization domain | RING-finger containing E3 ubiqutin ligase |
| Q9UNE7 | CHIP | C-terminus of Hsc70-interacting protein | 303 | DNA binding domain in p53, Hsp70 and CHIP complex * | U-box-containing E3 ubiqutin ligase |
| E3 Ubiquitin Ligase | Human Tumor Type | P53 Status |
|---|---|---|
| Mmd2 | Dominantly in soft tissue tumors, osteosarcomas, breast, prostate, colon cancers and esophageal carcinomas | Primarily Wild-type |
| Cop1 | Breast adenocarcinomas, ovarian adenocarcinomas, hepatocellular carcinoma (HCC), pancreatic cancer and colorectal cancer | Primarily Wild-type |
| Pirh2 | Hepatocellular carcinoma, clear cell renal cell carcinoma, lung neoplasms | Primarily Wild-type |
| CHIP | Non-small cell lung cancer and pancreatic cancer | Primarily mutant forms in tumors |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sane, S.; Rezvani, K. Essential Roles of E3 Ubiquitin Ligases in p53 Regulation. Int. J. Mol. Sci. 2017, 18, 442. https://doi.org/10.3390/ijms18020442
Sane S, Rezvani K. Essential Roles of E3 Ubiquitin Ligases in p53 Regulation. International Journal of Molecular Sciences. 2017; 18(2):442. https://doi.org/10.3390/ijms18020442
Chicago/Turabian StyleSane, Sanam, and Khosrow Rezvani. 2017. "Essential Roles of E3 Ubiquitin Ligases in p53 Regulation" International Journal of Molecular Sciences 18, no. 2: 442. https://doi.org/10.3390/ijms18020442
APA StyleSane, S., & Rezvani, K. (2017). Essential Roles of E3 Ubiquitin Ligases in p53 Regulation. International Journal of Molecular Sciences, 18(2), 442. https://doi.org/10.3390/ijms18020442