Human Immunodeficiency Virus Proteins Mimic Human T Cell Receptors Inducing Cross-Reactive Antibodies


Abstract
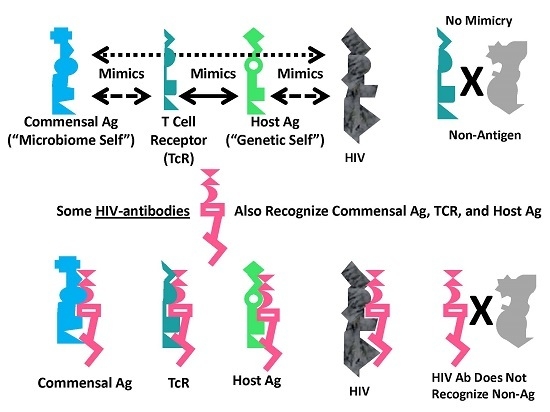
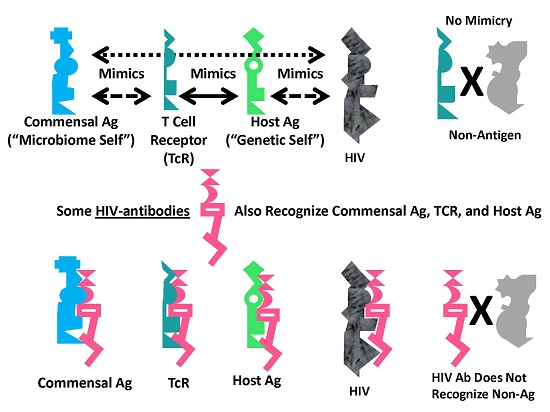
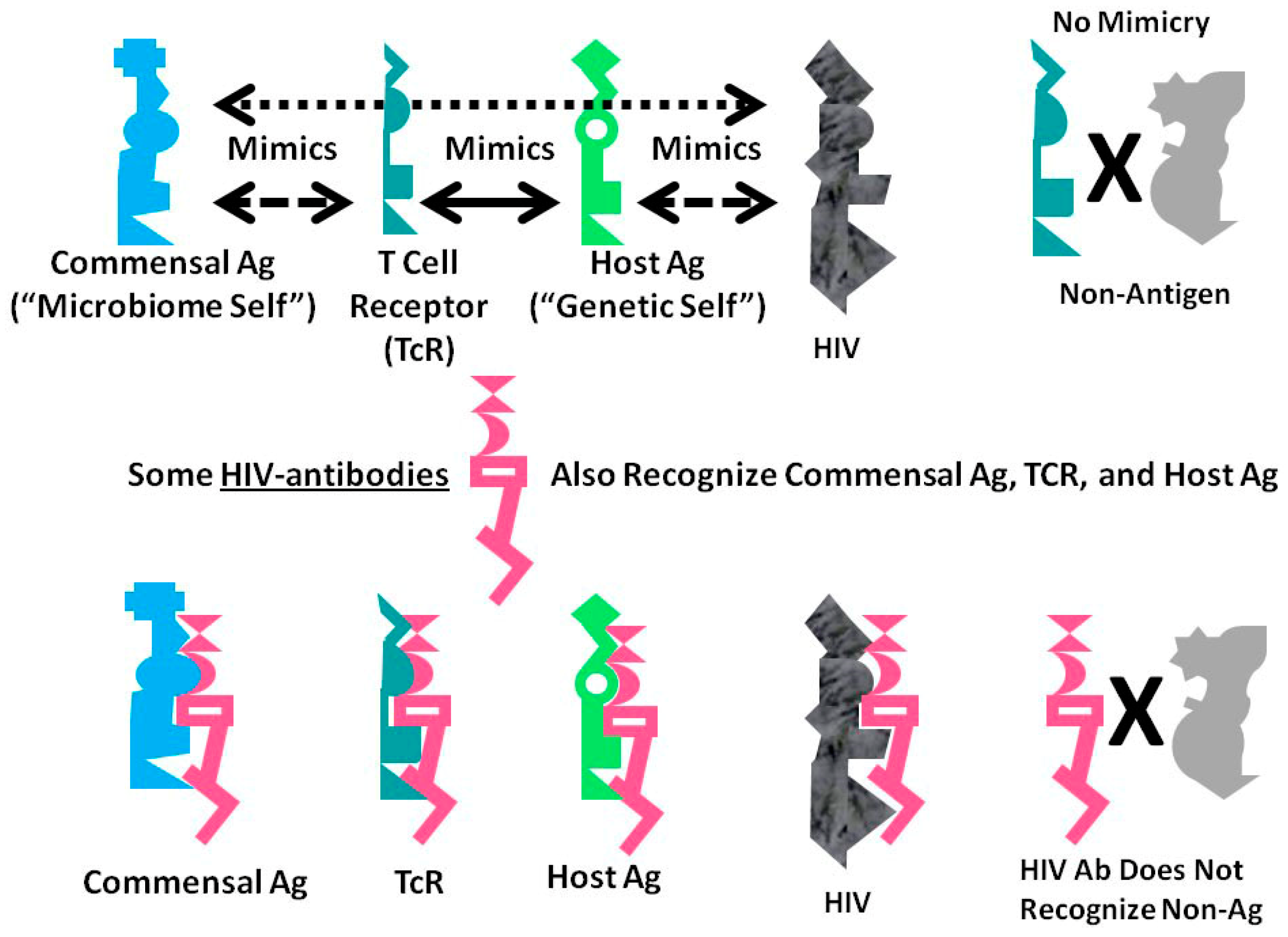
:
1. Introduction
2. Results
A Summary Model for HIV-Host-Microbiome Holoimmunity
3. Discussion
4. Methods
4.1. Proteonomics
4.2. Statistics
4.3. T Cell Receptor Peptide Synthesis
4.4. Antibodies
4.5. ELISA
5. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Margulis, L.; Fester, R. Symbiosis as a Source of Evolutionary Innovation; MIT Press: Cambridge, MA, USA, 1991. [Google Scholar]
- Mindell, D.P. Phylogenetic consequences of symbioses: Eukarya and Eubacteria are not monophyletic taxa. BioSystems 1992, 27, 53–62. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S. Autoimmunity and the microbiome: T-cell receptor mimicry of “self” and microbial antigens mediates self tolerance in holobionts. BioEssays 2016, 38, 1068–1083. [Google Scholar] [CrossRef] [PubMed]
- De Groot, A.S.; Moise, L.; Liu, R.; Gutierrez, A.H.; Tassone, R.; Bailey-Kellogg, C.; Martin, W. Immune camouflage: Relevance to vaccines and human immunology. Hum. Vaccin. Immunother. 2014, 10, 3570–3575. [Google Scholar] [CrossRef] [PubMed]
- Moise, L.; Terry, F.; Gutierrez, A.H.; Tassone, R.; Losikoff, P.; Gregory, S.H.; Bailey-Kellogg, C.; Martin, W.D.; de Groot, A.S. Smarter vaccine design will circumvent regulatory T cell-mediated evasion in chronic HIV and HCV infection. Front. Microbiol. 2014, 5, 502. [Google Scholar] [CrossRef] [PubMed]
- Moise, L.; Beseme, S.; Tassone, R.; Liu, R.; Kibria, F.; Terry, F.; Martin, W.; de Groot, A.S. T cell epitope redundancy: Cross-conservation of the TCR face between pathogens and self and its implications for vaccines and autoimmunity. Expert Rev. Vaccines 2016, 15, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Lieberman, J. Gaining a foothold: How HIV avoids innate immune recognition. Curr. Opin. Immunol. 2011, 23, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Rasaiyaah, J.; Tan, C.P.; Fletcher, A.J.; Price, A.J.; Blondeau, C.; Hilditch, L.; Jacques, D.A.; Selwood, D.L.; James, L.C.; Noursadeghi, M.; et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 2013, 503, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Guha, D.; Ayyavoo, V. Innate immune evasion strategies by Human Immunodeficiency Virus Type 1. ISRN AIDS 2013, 954806. [Google Scholar] [CrossRef] [PubMed]
- Via, C.S.; Shearer, G.M. Autoimmunity and the acquired immune deficiency syndrome. Curr. Opin. Immunol. 1989, 1, 753–756. [Google Scholar] [CrossRef]
- Malatzky-Goshen, E.; Shoenfeld, Y. AIDS and autoimmunity. Autoimmunity 1989, 3, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Morrow, W.J.; Isenberg, D.A.; Sobol, R.E.; Stricker, R.B.; Kieber-Emmons, T. AIDS virus infection and autoimmunity: A perspective of the clinical, immunological, and molecular origins of the autoallergic pathologies associated with HIV disease. Clin. Immunol. Immunopathol. 1991, 58, 163–180. [Google Scholar] [CrossRef]
- Bjork, R.L. HIV-1: Seven facets of functional molecular mimicry. Immunol. Lett. 1991, 28, 91–96. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S.; Hobbs, S.H. Homologies between mycoplasma adhesion peptide, CD4, and class II MHC proteins: A possible mechanism for HIV mycoplasma synergism in AIDS. Res. Immunol. 1992, 142, 519–523. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S. Rethinking AIDS. The Tragic Cost of Premature Concensus; Free Press: New York, NY, USA, 1983. [Google Scholar]
- Russo, S.; Lopalco, L. Is autoimmunity a component of natural immunity to HIV? Curr. HIV Res. 2006, 4, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.M.; Hong, X.Z.; Xu, J.H.; Luo, J.X.; Mo, H.Y.; Zhao, H.L. Autoimmunity and dysmetabolism of human acquired immunodeficiency syndrome. Immunol. Res. 2016, 64, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Beretta, A.; Grassi, F.; Pelagi, M.; Clivio, A.; Parravicini, C.; Giovinazzo, G.; Andronico, F.; Lopalco, L.; Verani, P.; Buttò, S. HIV env glycoprotein shares a cross-reacting epitope with a surface protein present on activated human monocytes and involved in antigen presentation. Eur. J. Immunol. 1987, 17, 1793–1798. [Google Scholar] [CrossRef] [PubMed]
- Golding, H.; Robey, F.A.; Gates, F.T.; Linder, W.; Beining, P.R.; Hoffman, T.; Golding, B. Identification of homologous regions in human immunodeficiency virus I gp41 and human MHC class II β 1 domain. I. Monoclonal antibodies against the gp41-derived peptide and patients' sera react with native HLA class II antigens, suggesting a role for autoimmunity in the pathogenesis of acquired immune deficiency syndrome. J. Exp. Med. 1988, 167, 914–923. [Google Scholar] [PubMed]
- Golding, H.; Shearer, G.M.; Hillman, K.; Lucas, P.; Manischewitz, J.; Zajac, R.A.; Clerici, M.; Gress, R.E.; Boswell, R.N.; Golding, B. Common epitope in human immunodeficiency virus (HIV) I-GP41 and HLA class II elicits immunosuppressive autoantibodies capable of contributing to immune dysfunction in HIV I-infected individuals. J. Clin. Invest. 1989, 83, 1430–1435. [Google Scholar] [CrossRef] [PubMed]
- Grassi, F.; Meneveri, R.; Gullberg, M.; Lopalco, L.; Rossi, G.B.; Lanza, P.; De Santis, C.; Brattsand, G.; Buttò, S.; Ginelli, E. Human immunodeficiency virus type 1 gp120 mimics a hidden monomorphic epitope borne by class I major histocompatibility complex heavy chains. J. Exp. Med. 1991, 174, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Lopalco, L.; de Santis, C.; Meneveri, R.; Longhi, R.; Ginelli, E.; Grassi, F.; Siccardi, A.G.; Beretta, A. Human immunodeficiency virus type 1 gp120 C5 region mimics the HLA class I alpha 1 peptide-binding domain. Eur. J. Immunol. 1993, 23, 2016–2021. [Google Scholar] [CrossRef] [PubMed]
- Powell, P.D.; DeMartini, J.C.; Azari, P.; Stargell, L.A.; Cordain, L.; Tucker, A. Evolutionary stable strategy: A test for theories of retroviral pathology which are based upon the concept of molecular mimicry. J. Theor. Biol. 2000, 202, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S.; Dewitt, S.H. CD4 similarity to proteins of infectious agents in AIDS and their role in autoimmunity. Med. Hypoth. 1994, 43, 361–371. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S. Preliminary evidence of idiotype-antiidiotype immune complexes cross reactive with lymphocyte antigens in AIDS and lupus. Med. Hypoth. 1995, 44, 20–27. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S. Antigenic complementarity between HIV and other AIDS-associated infections results in idiotype-antiidiotype antibody complexes that cross-react with lymphocyte proteins. Vaccine 2005, 23, 2160–2163. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S.; Rallo, A. Antigenic complementarity resulting in idiotype-antiidiotype immune complexes: Possible contributor to AIDS pathogenesis and autoimmunity. Autoimmunity 2004, 37, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S. Antigenic complementarity among AIDS-associated infectious agents and molecular mimicry of lympohocyte proteins as inducers of lymphocytotoxic antibodies and circulating immune complexes. J. Clin. Virol. 2004, 31, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.R.; Rogers, R.A.; Brain, J.D. Shared antigenic epitopes on the V3 loop of HIV-1 gp120 and proteins on activated human T cells. Virology 1998, 246, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Lake, D.F.; Schlute, S.F.; Wang, E.; Bernstein, R.M.; Edmundson, A.B.; Marchalonis, J.J. Autoantibodies to the α/β T-cell receptors in human immunodeficiency virus infection: dysregulation and mimicry. Proc. Natl. Acad. Sci. USA 1994, 91, 10849–10853. [Google Scholar] [CrossRef] [PubMed]
- Marchalonis, J.J.; Lake, D.F.; Schluter, S.F.; Dehghanpisheh, K.; Watson, R.R.; Ampel, N.M.; Galgiani, J.N. Autoantibodies against peptide-defined epitopes of T-cell receptors in retrovirally infected humans and mice. Adv. Exp. Med. Biol. 1995, 383, 211–222. [Google Scholar] [PubMed]
- Sheikh, J.; Ongradi, J.; Austen, B.; Dalgleish, A. The potential importance of MHC mimicry by HIV in the pathogenesis of AIDS. Biochem. Soc. Trans. 1995, 23, 471–479. [Google Scholar] [CrossRef]
- Süsal, C.; Kröpelin, M.; Daniel, V.; Opelz, G. Molecular mimicry between HIV-1 and antigen receptor molecules: A clue to the pathogenesis of AIDS. Vox Sang. 1993, 65, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Dalgleish, A.G. Autoimmune mechanisms of depletion of CD4 cells in HIV infection. Br. J. Haematol. 1995, 91, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Xiao, Y.; Dierich, M.P. HIV-1 gp41 and type I interferon: Sequence homology and biological as well as clinical implications. Immunol. Res. 2000, 22, 61–66. [Google Scholar] [CrossRef]
- Serres, P.F. AIDS: An immune response against the immune system. Role of a precise tridimensional molecular mimicry. J. Autoimmun. 2001, 16, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Sanhadji, K.; Tardy, J.C.; Touraine, J.L. HIV-1 infection: Functional competition between gp41 and interleukin-2. C. R. Biol. 2010, 333, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Min, W.; Taylor, E.W. An HIV-1 encoded peptide mimics the DNA binding loop of NF-κB and binds thioredoxin with high affinity. Mutat. Res. 2005, 579, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Bettaieb, A.; Fromont, P.; Louache, F.; Oksenhendler, E.; Vainchenker, W.; Duédari, N.; Bierling, P. Presence of cross-reactive antibody between human immunodeficiency virus (HIV) and platelet glycoproteins in HIV-related immune thrombocytopenic purpura. Blood 1992, 80, 162–169. [Google Scholar] [PubMed]
- Dominguez, V.; Gevorkian, G.; Govezensky, T.; Rodriguez, H.; Viveros, M.; Cocho, G.; Macotela, Y.; Masso, F.; Pacheco, M.; Estrada, J.L.; et al. Antigenic homology of HIV-1 GP41 and human platelet glycoprotein GPIIIa (integrin beta3). J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1998, 17, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Tishkoff, G.H.; Hunt, L.T. Unexpected molecular mimicry among peptides MHC class II, blood-clotting factor X, and HIV-1 envelope glycoprotein GP120. Thromb. Res. 2000, 98, 343–346. [Google Scholar] [CrossRef]
- Tsiakalos, A.; Routsias, J.G.; Kordossis, T.; Moutsopoulos, H.M.; Tzioufas, A.G.; Sipsas, N.V. Fine epitope specificity of anti-erythropoietin antibodies reveals molecular mimicry with HIV-1 p17 protein: A pathogenetic mechanism for HIV-1-related anemia. J. Infect. Dis. 2011, 204, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Nardi, M.A.; Karpatkin, S. Role of molecular mimicry to HIV-1 peptides in HIV-1-related immunologic thrombocytopenia. Blood 2005, 106, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Pintér, C.; Siccardi, A.G.; Lopalco, L.; Longhi, R.; Clivio, A. HIV glycoprotein 41 and complement factor H interact with each other and share functional as well as antigenic homology. AIDS Res. Hum. Retroviruses 1995, 11, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Wiwanitkit, V. Structural homology of HIV-1 GP41 and human platelet glycoprotein GPIIIa. Blood Coagul. Fibrinolysis 2008, 19, 739–740. [Google Scholar] [CrossRef] [PubMed]
- Witte, V.; Laffert, B.; Rosorius, O.; Lischka, P.; Blume, K.; Galler, G.; Stilper, A.; Willbold, D.; D’Aloja, P.; Sixt, M.; et al. HIV-1 Nef mimics an integrin receptor signal that recruits the polycomb group protein Eed to the plasma membrane. Mol. Cell. 2004, 13, 179–190. [Google Scholar] [CrossRef]
- Pornillos, O.; Higginson, D.S.; Stray, K.M.; Fisher, R.D.; Garrus, J.E.; Payne, M.; He, G.P.; Wang, H.E.; Morham, S.G.; Sundquist, W.I. HIV Gag mimics the Tsg101-recruiting activity of the human Hrs protein. J. Cell Biol. 2003, 162, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Spehar, T.; Strand, M. Cross-reactivity of anti-human immunodeficiency virus type 1 gp41 antibodies with human astrocytes and astrocytoma cell lines. J. Virol. 1994, 68, 6262–6269. [Google Scholar] [PubMed]
- Spehar, T.; Strand, M. Molecular mimicry between HIV-1 gp41 and an astrocyte isoform of α-actinin. J. Neurovirol. 1995, 1, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Zurbriggen, A.; Oldstone, M.B.; Fujinami, R.S. Common immunologic determinant between human immunodeficiency virus type 1 gp41 and astrocytes. J. Virol. 1991, 65, 1370–1376. [Google Scholar] [PubMed]
- Trujillo, J.R.; McLane, M.F.; Lee, T.H.; Essex, M. Molecular mimicry between the human immunodeficiency virus type 1 gp120 V3 loop and human brain proteins. J. Virol. 1993, 67, 7711–7715. [Google Scholar] [PubMed]
- Eddleston, M.; de la Torre, J.C.; Xu, J.Y.; Dorfman, N.; Notkins, A.; Zolla-Pazner, S.; Oldstone, M.B. Molecular mimicry accompanying HIV-1 infection: Human monoclonal antibodies that bind to gp41 and to astrocytes. AIDS Res. Hum. Retroviruses 1993, 9, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Atassi, H.; Atassi, M.Z. HIV envelope proteinis recognized as an alloantigen by human DR-specific alloreactive T cells. Hum. Immunol. 1992, 34, 31–38. [Google Scholar] [CrossRef]
- Nikolich-Zugich, J.; Slifka, M.K.; Messaoudi, I. The many important facets of T-cell repertoire diversity. Nat. Rev. Immunol. 2004, 4, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Berson, S.A.; Yalow, R.S. Quantitative aspects of the reaction between insulin and insulin-binding antibody. J. Clin. Invest. 1959, 38, 1996–2016. [Google Scholar] [CrossRef] [PubMed]
- Burch, H.B.; Clement, S.; Sokol, M.S.; Landry, F. Reactive hypoglycemic coma due to insulin autoimmune syndrome: case report and literature review. Am. J. Med. 1992, 92, 681–685. [Google Scholar] [CrossRef]
- Suzuki, T.; Nishii, N.; Takashima, S.; Matsubara, T.; Iwasawa, A.; Takeuchi, H.; Tahara, K.; Hachisu, T.; Kitagawa, H. Ligand-binding characteristics of feline insulin-binding immunoglobulin G. J. Vet. Med. Sci. 2015, 77, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.L.; Fincke, J.E.; Sharer, L.R.; Monos, D.S.; Lu, S.; Gaughan, J.; Platsoucas, C.D.; Oleszak, E.L. Oligoclonal T cells are infiltrating the brains of children with AIDS: Sequence analysis reveals high proportions of identical β-chain T-cell receptor transcripts. Clin. Exp. Immunol. 2005, 338–356. [Google Scholar] [CrossRef] [PubMed]
- Norley, S.; Kurth, R. The role of the immune response during SIVagm infection of the African green monkey natural host. Front. Biosci. 2004, 9, 550–564. [Google Scholar] [PubMed]
- Ansari, A.A. Autoimmunity, anergy, lentiviral immunity and disease. Autoimmun. Rev. 2004, 3, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Lucchese, G.; Stufano, A.; Calabro, M.; Kanduc, D. Charting the peptide crossreactome between HIV-1 and the human proteome. Front. Biosci. 2011, 3, 1385–4000. [Google Scholar] [CrossRef]
- Venturi, V.; Price, D.A.; Douek, D.C.; Davenport, M.P. The molecular basis for public T-cell responses? Nat. Rev. Immunol. 2008, 8, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.J.; Douek, D.C.; Price, D.A. Bias in the alphabeta T-cell repertoire: Implications for disease pathogenesis and vaccination. Immunol. Cell Biol. 2011, 89, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Zvyagin, I.V.; Pogorelyy, M.V.; Ivanova, M.E.; Komech, E.A.; Shugay, M.; Bolotin, D.A.; Shelenkov, A.A.; Kurnosov, A.A.; Staroverov, D.B.; Chudakov, D.M.; et al. Distinctive properties of identical twins’ TCR repertoires revealed by high-throughput sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, 5980–5985. [Google Scholar] [CrossRef] [PubMed]
- Williams, W.B.; Liao, H.X.; Moody, M.A.; Kepler, T.B.; Alam, S.M.; Gao, F.; Wiehe, K.; Trama, A.M.; Jones, K.; Zhang, R.; et al. HIV-1 vaccines. Diversion of HIV-1 vaccine-induced immunity by gp41-microbiota cross-reactive antibodies. Science 2015, 349, aab1253. [Google Scholar] [CrossRef] [PubMed]
- Rolland, M.; Nickle, D.C.; Deng, W.; Frahm, N.; Brander, C.; Learn, G.H.; Heckerman, D.; Jojic, N.; Jojic, V.; Walker, B.D.; et al. Recognition of HIV-1 peptides by host CTL is related to HIV-1 similarity to human proteins. PLoS ONE 2007, 2, e823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Kuyl, A.C. HIV infection and HERV expression: A review. Retrovirology 2012, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Seifarth, W.; Frank, O.; Zeilfelder, U.; Spiess, B.; Greenwood, A.D.; Hehlmann, R.; Leib-Mösch, C. Comprehensive analysis of human endogenous retrovirus transcriptional activity in human tissues with a retrovirus-specific microarray. J. Virol. 2005, 79, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.N.; Carnegie, P.R.; Martin, J.; Davari Ejtehadi, H.; Hooley, P.; Roden, D.; Rowland-Jones, S.; Warren, P.; Astley, J.; Murray, P.G. Demystified. Human endogenous retroviruses. Mol. Pathol. 2003, 6, 11–18. [Google Scholar] [CrossRef]
- Trama, A.M.; Moody, M.A.; Alam, S.M.; Jaeger, F.H.; Lockwood, B.; Parks, R.; Lloyd, K.E.; Stolarchuk, C.; Scearce, R.; Foulger, A.; et al. HIV-1 envelope gp41 antibodies can originate from terminal ileum B cells that share cross-reactivity with commensal bacteria. Cell Host Microbe 2014, 16, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Borgdorff, H.; Tsivtsivadze, E.; Verhelst, R.; Marzorati, M.; Jurriaans, S.; Ndayisaba, G.F.; Schuren, F.H.; van de Wijgert, J.H. Lactobacillus-dominated cervicovaginal microbiota associated with reduced HIV/STI prevalence and genital HIV viral load in African women. ISME J. 2014, 8, 1781–1793. [Google Scholar] [CrossRef] [PubMed]
- Hearps, A.C.; Tyssen, D.; Srbinovski, D.; Bayigga, L.; Diaz, D.J.; Aldunate, M.; Cone, R.A.; Gugasyan, R.; Anderson, D.J.; Tachedjian, G. Vaginal lactic acid elicits an anti-inflammatory response from human cervicovaginal epithelial cells and inhibits production of pro-inflammatory mediators associated with HIV acquisition. Mucosal Immunol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Eade, C.R.; Diaz, C.; Chen, S.; Cole, A.L.; Cole, A.M. HIV-Enhancing factors are secreted by reproductive epithelia upon inoculation with bacterial vaginosis-associated bacteria. Protein Pept. Lett. 2015, 22, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Van de Wijgert, J.H.; Jespers, V. The global health impact of vaginal dysbiosis. Res. Microbiol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Zabihollahi, R.; Motevaseli, E.; Sadat, S.M.; Azizi-Saraji, A.R.; Asaadi-Dalaie, S.; Modarressi, M.H. Inhibition of HIV and HSV infection by vaginal lactobacilli in vitro and in vivo. DARU J. Pharm. Sci. 2012, 20, 53. [Google Scholar] [CrossRef] [PubMed]
- Mirmonsef, P.; Spear, G.T. The barrier to HIV transmission provided by genital tract Lactobacillus colonization. Am. J. Reprod. Immunol. 2014, 1, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Petrova, M.I.; van den Broek, M.; Balzarini, J.; Vanderleyden, J.; Lebeer, S. Vaginal microbiota and its role in HIV transmission and infection. FEMS Microbiol. Rev. 2013, 37, 762–792. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, J.L.; Stites, D.P. Hypothesis: AIDS is an autoimmune disease directed at the immune system and triggered by a lymphotropic retrovirus. Clin. Immunol. Immunopathol. 1986, 41, 305–313. [Google Scholar] [CrossRef]
- Stott, E.J. Anti-cell antibody in macaques. Nature 1991, 353, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Kion, T.A.; Hoffmann, G.W. Anti-HIV and anti-anti-MHC antibodies in alloimmune and autoimmune mice. Science 1991, 253, 1138–1140. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.W.; Kion, T.A.; Grant, M.D. An idiotypic network model of AIDS immunopathogenesis. Proc. Natl. Acad. Sci. USA 1991, 88, 3060–3064. [Google Scholar] [CrossRef] [PubMed]
- Schacker, T.W.; Reilly, C.; Haase, A.T. Collagen deposition in HIV-1 infected lymphatic tissues and T cell homeostasis. J. Clin. Invest. 2002, 110, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Lopalco, L.; Burastero, S.E. HIV-1 and the self-nonself connection: How to sleep with the enemy and be much better off. AIDS Rev. 2008, 10, 162–171. [Google Scholar] [PubMed]
- Stratov, I.; DeRose, R.; Purcell, D.F.; Kent, S.J. Vaccines and vaccine strategies against HIV. Curr. Drug Targets 2004, 5, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Ersching, J.; Pinto, A.R. HIV-1 vaccine clinical trials: the Brazilian experience. Rev. Med. Virol. 2009, 19, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S. Five myths about AIDS that are misdirecting research and treatment. Genetica 1995, 95, 100–132. [Google Scholar] [CrossRef]
- Keay, S.; Tacket, C.O.; Murphy, J.R.; Handwerger, B.S. Anti-CD4 anti-idiotype antibodies in volunteers immunized with rgpl60 or HIV-1 or infected with HIV-1. AIDS Res. Human Retroviruses 1992, 8, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- De Santis, C.; Robbioni, P.; Longhi, R.; Lopalco, L.; Siccardi, A.G.; Beretta, A.; Roberts, N.J. Cross-reactive response to human immunodeficiency virus type 1 (HIV-1) gp120 and HLA class I heavy chains induced by receipt of HIV-1-derived envelope vaccines. J. Infect. Dis. 1993, 168, 1396–1403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Verkoczy, L.; Wiehe, K.; Alam, S.M.; Nicely, N.I.; Santra, S.; Bradley, T.; Pemble, C.W.; Zhang, J.; Gao, F.; Monefiori, D.C.; et al. Initiation of immune tolerance–controlled HIV gp41 neutralizing B cell lineages. Sci. Trans. Med. 2016, 8, 336ra62. [Google Scholar] [CrossRef] [PubMed]
- Moody, M.A.; Pedroza-Pacheco, I.; Vandergrift, N.A.; Chui, C.; Lloyd, K.E.; Parks, R.; Soderberg, K.A.; Ogbe, A.T.; Cohen, M.S.; Liao, H.-X.; Gao, F.; et al. Immune perturbations in HIV-1–infected individuals who make broadly neutralizing antibodies. Sci. Immun. 2016, 1, aag0851. [Google Scholar] [CrossRef] [PubMed]
- Bradac, J.; Dieffenbach, C.W. HIV vaccine development: Lessons from the past, informing the future. Invest. Drugs J. 2009, 12, 435–439. [Google Scholar]
- Shearer, G.M.; Pinto, L.A.; Clerici, M. Alloimmunization for immune-based therapy and vaccine design against HIV/AIDS. Immunol. Today 1999, 20, 66–71. [Google Scholar] [CrossRef]
- Bourinbaiar, A.; Root-Bernstein, R.S.; Abulafia-Lapid, R.; Rytik, P.G.; Kanev, A.N.; Orlovsky, V.G. Therapeutic HIV vaccines. Curr. Pharm. Design 2006, 12, 2017–2030. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S.; Bernstein, M. SIV as a human vaccine against HIV? J. Theor. Biol. 1996, 180, 91–92. [Google Scholar] [CrossRef]
- Kou, Z.C.; Puhr, J.S.; Wu, S.S.; Goodenow, M.M.; Sleasman, J.W. Combination antiretroviral therapy results in a rapid increase in T cell receptor variable region β repertoire diversity within CD45RA CD8 T cells in human immunodeficiency virus-infected children. J. Infect. Dis. 2003, 187, 385–395. [Google Scholar] [CrossRef] [PubMed]
- McFarland, E.J.; Harding, P.A.; Striebich, C.C.; McWhinney, S.; Kuritzkes, D.R.; Kotzin, B.L. Clonal CD8+ T Cell expansions in peripheral blood from human immunodeficiency virus type 1-infected children. J. Infect. Dis. 2002, 186, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Weekes, M.P.; Carmichael, A.J.; Wills, M.R.; Mynard, K.; Sissons, J.G.P. Human CD38-CD8+ T cells contain greatly expanded functional virus-specific memory CTL clones. J. Immunol. 1999, 162, 7569–7577. [Google Scholar] [PubMed]
- Rudensky, A.Y.; Preston-Hurlburt, P.; Hong, S.C.; Barlow, A. Sequence analysis of peptides bound to MHC class II molecules. Nature 1991, 353, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.W.; McCormack, J.M.; Fenderson, P.G.; Ho, M.K. Human and murine antibodies cross-reactive with streptococcal M protein and myosin recognize the sequence GLN-LYS-SER-LYS-GLN in M protein. J. Immunol. 1989, 143, 2677–2683. [Google Scholar] [PubMed]
- Root-Bernstein, R.S.; Podufaly, A. Autoreactive T-cell receptor (Vβ/D/Jβ) sequences in diabetes recognize insulin, the insulin receptor, and each other, and are targets of insulin antibodies. Open Autoimmun. J. 2012, 4, 10–22. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S. Rethinking molecular mimicry in rheumatic heart disease and autoimmune myocarditis: Laminin, collagen IV, CAR, and B1AR as initial targets of disease. Front. Ped. Rheumatol. 2014, 2, 85. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, S.; Robert-Guroff, M.; Rusche, J.; Koito, A.; Hattori, T.; Hoshino, H.; Javaherian, K.; Takatsuki, K.; Putney, S. Characterization of a Human immunodeficiency virus neutralizing monoclonal antibody and mapping of the neutralizing epitope. J. Virol. 1998, 62, 2107–2114. [Google Scholar]
- Hauber, J.; Perkins, A.; Heimer, E.; Cullen, B. Trans-activation of human immunodeficiency virus gene expression is mediated by nuclear events. Proc. Natl. Acad. Sci. USA 1987, 84, 6364–6368. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, L.; Mullenax, J.; Nelson, R.; Merigan, T. Viral polypeptides detected by a complement-dependent neutralizing murine monoclonal antibody to human cytomegalovirus. J. Virol. 1985, 55, 274–280. [Google Scholar] [PubMed]
- Shugars, D.C.; Smith, M.S.; Glueck, D.H.; Nantermet, P.V.; Seillier-Moiseiwitsch, F.; Swanstrom, R. Analysis of human immunodeficiency virus type 1 nef gene sequences present in vivo. J. Virol. 1993, 67, 4639–4650. [Google Scholar] [PubMed]
- Hagen, M.D.; Meyer, K.B.; Kopelman, R.I.; Pauker, S.G. Human immunodeficiency virus infection in health care workers. A method for estimating individual occupational risk. Arch. Intern. Med. 1989, 49, 1541–1544. [Google Scholar] [CrossRef]
- Boily, M.C.; Baggaley, R.F.; Wang, L.; Masse, B.; White, R.G.; Hayes, R.J. Heterosexual risk of HIV-1 infection per sexual act: Systematic review and meta-analysis of observational studies. Lancet Infect. Dis. 2009, 9, 118–129. [Google Scholar] [CrossRef]
- Sonnabend, J.A.; Saadoun, S. The acquired immunodeficiency syndrome: A discussion of etiologic hypotheses. AIDS Res. 1983, 1, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Sonnabend, J.A.; Witkin, S.S.; Purtilo, D.T. A multifactorial model for the development of AIDS in homosexual men. Ann. N. Y. Acad. Sci. 1984, 437, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S. Do we know the cause(s) of AIDS? Persp. Biol. Med. 1990, 33, 480–500. [Google Scholar] [CrossRef]
- Pattacini, L.; Baeten, J.M.; Thomas, K.K.; Fluharty, T.R.; Murnane, P.M.; Donnell, D.; Bukusi, E.; Ronald, A.; Mugo, N.; Lingappa, J.R.; et al. Regulatory T-cell activity but not conventional HIV-specific T-Cell responses are associated with protection from HIV-1 infection. J. Acquir. Immune Defic. Syndr. 2016, 72, 119–128. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Virus | TCR AVG% N = 386 | Chi 2 p Val HIV vs. AVG | HIV TCR% N = 109 | Chi 2 p Val HIV vs. Rand | Rand + Anti TCR% N = 201 | Chi 2 p Val AVG vs. Rand |
|---|---|---|---|---|---|---|
| Adenovirus | 14 | 16 | 15.5 | |||
| Astrovirus | 1 | 2 | 2.5 | |||
| Bocavirus | 0.5 | 2 | 4 | |||
| Cardiovirus | 0.5 | 0 | 1.5 | |||
| Coronavirus | 5 | 9 | 4 | |||
| Coxsackie A | 5 | 0.001 | 12 | 1.3 × 10−7 | 3 | |
| Coxsackie B | 3.3 | 4 | 0 | |||
| CMV | 22.5 | 30 | 3.1 × 10−6 | 13.5 | ||
| Echoviruses | 7.2 | 6 | 2.5 | |||
| Enteroviruses | 7.3 | 14 | 3.3 × 10−7 | 4 | ||
| EBV | 8.1 | 0.003 | 16 | 4.1 × 10−6 | 5.5 | |
| HAV | 0.8 | 4 | 0 | |||
| HBV | 7 | 1.6 × 10−5 | 18 | 21.5 | 1 × 10−8 | |
| HCV | 21.5 | 7.3 × 10−5 | 41 | 33 | ||
| HEV | 2.7 | 5 | 14 | 0.001 | ||
| HHV1 | 4.5 | 7 | 14.5 | 0.004 | ||
| HHV2 | 4.5 | 0.001 | 11 | 7.5 | ||
| HHV6 | 3.7 | 8.4 × 10−7 | 13 | 8.5 | ||
| HHV8 | 3.7 | 8 | 8.5 | |||
| HIV-1 | 71.2 | 2.6 × 10−6 | 87 | 6 × 10−8 | 60.5 | |
| HTLV I and II | 2.7 | 2 | 6 | |||
| Infl A Virus | 20.8 | 27 | 29 | |||
| Infl B virus | 1.3 | 1 | 5.5 | |||
| Infl C virus | 0.7 | 1 | 1 | |||
| Jap enc virus | 1.5 | 3 | 0.5 | |||
| Measles virus | 2.8 | 2 | 8 | |||
| Mumps virus | 0.5 | 0 | 3 | |||
| Norovirus | 5.3 | 8 | 5.5 | |||
| Papilloma virus | 15.5 | 0.003 | 29 | 33.5 | 0.0001 | |
| Parainfluenza | 0.7 | 4 | 3 | |||
| Polio virus | 0.5 | 4 | 1 | |||
| Polyoma virus | 2 | 1 | 1.5 | |||
| Reovirus | 2.3 | 3 | 2.5 | |||
| RSV | 0.5 | 2 | 7.5 | 0.007 | ||
| Rhinovirus | 4 | 9 | 5.5 | |||
| Rotaviruses | 6.3 | 1 × 10−5 | 17 | 14.5 | 0.0007 | |
| Rubella | 1.3 | 2 | 4 | |||
| Varicella zoster | 4.3 | 6 | 4 |
| Human Microbe | AVG TCR% N = 386 | Chi Sq p Val AVG vs. HIV | HIV TCR% N = 109 | Chi Sq p Val Rand vs. HIV | Rand + Anti TCR % N = 201 | Chi Sq p Val AVG vs. Rand |
|---|---|---|---|---|---|---|
| Bacillus cereus | 32.0 | 37 | 42.5 | |||
| Bacteroides spp. | 64.0 | 70 | 74 | |||
| Bifidobacteria | 27.5 | 0.005 | 40 | 42.5 | 0.0008 | |
| Bordetella pertussis | 5.0 | 9 | 16.5 | 0.002 | ||
| Campylobacter jejuni | 4.8 | 5 | 6 | |||
| Candida albicans | 3.9 | 3 × 10−6 | 13 | 10 | 0.0002 | |
| ardiobacteria | 0.5 | 7 × 10−6 | 4 | 8 | <1 × 10−10 | |
| Chlamydia | 5.3 | <1 × 10−10 | 26 | <1 × 10−10 | 1 | |
| Clostridium spp. | 50.5 | 0.007 | 64 | 61.5 | ||
| Coccidiodes spp. | 39.5 | 33 | 26 | |||
| Coprococcus | 10.5 | 14 | 7.5 | |||
| Corynebacteria | 14.0 | <1 × 10−10 | 43 | 53.5 | <1 × 10−10 | |
| Cryptococcus neoformans | 1.0 | <1 × 10−10 | 24 | 18.5 | <1 × 10−10 | |
| Cryptosporidium | 15.5 | 13 | 18 | |||
| Entamoeba | 6.7 | 1 × 10−8 | 21 | 24.5 | <1 × 10−10 | |
| Enterobacter spp. | 12.0 | <1 × 10−10 | 46 | 0.002 | 61 | <1 × 10−10 |
| Enterococcus spp. | 22.3 | 6.6 × 10−7 | 43 | 0.0002 | 26.5 | |
| Escherichia coli | 26.8 | 36 | 0.0005 | 53.5 | 9 × 10−8 | |
| Eubacterium | 22.0 | 0.0007 | 36 | 47.5 | 3.3 × 10−7 | |
| Gardnerella vaginalis | 13.5 | 9 | 11 | |||
| Giardia | 11.5 | 0.0003 | 23 | 27 | 1.2 × 10−6 | |
| Haemophilus influenzae | 4.6 | 6 | 4 | |||
| Helicobacter pyelori | 7.6 | 8 | 7.5 | |||
| Histoplasmosis | 0 | 0 | 1 | |||
| Isospora | 0 | 0 | 0 | |||
| Klebsiella pneumoniae | 10.9 | 17 | 23.5 | |||
| Lactobacillus spp. | 40.0 | 45 | 0.0001 | 63.5 | 1 × 10−6 | |
| Legionella pneumophila | 6.0 | 6 | 11 | |||
| Listeria | 8.9 | 8 | 0.008 | 16 | ||
| M. tuberculosis | 8.3 | 0.0004 | 18 | 29 | 5 × 10−6 | |
| Atypical Mycobacterium | 35.0 | <1 × 10−10 | 74 | 76.5 | <1 × 10−10 | |
| Mycoplasma | 9.0 | 8 | 13.5 | |||
| Neisseria | 6.2 | 0.0001 | 22 | 20.3 | 0.0005 | |
| Pneumocystis | 9 | 6 | 13 | |||
| Prevotella spp. | 47.5 | 50 | 50.5 | |||
| Pseudomona aeruginosa | 11.6* | 0.002 | 21 | 31.5 | 0.0003 | |
| Salmonella | 10.5 | <1 × 10−10 | 29 | 29 | <1 × 10−10 | |
| Shigella dysenteriae | 4.0 | 3 | 5.5 | |||
| Staphylococcus | 12.5 | <1 × 10−10 | 34 | 27.5 | 0.0008 | |
| Streptococcus spp. | 31.0 | <1 × 10−10 | 73 | 80.5 | <1 × 10−10 | |
| Toxoplasma gondii | 25.0 | 3 × 10−5 | 43 | 38 | 0.003 | |
| Treponema pallidum | 1.3 | 3 | 0 | |||
| Trichomonas vaginalis | 15.0 | 2 × 10−8 | 35 | 33 | 4.6 × 10−7 | |
| Trypanosoma cruzi | 14.0 | <1 × 10−10 | 36 | 31 | 9.6 × 10−7 |
| HIV-1 Proteins | % TCR Normal Mimics | % TCR Control Mimics | % TCR Antisense Control Mimics | % TCR Random Sequence Mimics | % TCR HIV Mimics |
|---|---|---|---|---|---|
| Env (envelope proteins, gp160, gp120, gp41) | 70 | 63 | 27 | 40 | 69 |
| Pol (reverse transcriptase, protease, RNAase, integrase) | 30 | 20 | 22 | 21 | 20 |
| Gag (viral capsid and matrix, p24, p17, p9, p6) | 20 | 6 | 18 | 15 | 30 |
| Nef (regulatory: negative replication factor) | 11 | 9 | 17 | 11 | 0 |
| Vif (regulatory: virion infectivity factor) | 2 | 4 | 8 | 7 | 0 |
| Vpu (regulatory: viral protein U, virus assembly) | 3 | 3 | 1 | 6 | 3 |
| Tat (regulatory: transactivator of RNA transcription) | 5 | 2 | 6 | 4 | 0 |
| Rev (regulatory: stimulates protein production) | 7 | 2 | 9 | 6 | 13 |
| Vpr (regulatory: viral protein R, protein production accelerator) | 0 | 1 | 3 | 3 | 3 |
| Total HIV mimics | 148 | 110 | 111 | 113 | 145 |
| One or more HIV mimics | 79 | 76 | 61 | 60 | 80 |
| No HIV mimics | 21 | 24 | 39 | 40 | 20 |
| Average Total BLAST Human Similarities per HIV TCR | Average HIV TCR Similarities to Other Human TCR/Ig per HIV TCR | Average Non-TCR/Ig Human Similarities per HIV TCR |
|---|---|---|
| 28.5 ± 19.0 | 10.7 ± 10.7 | 17.5 ± 16.5 |
| TCR ID | Sequence | Patient |
|---|---|---|
| HIV 1 | CASSEELAGGSYNE | NP95-73 |
| HIV 2 | CASSERGTNSPL | NP95-184-O |
| HIV 3 | CASSLELAKNI | NP95-184-O |
| HIV 4 | CASSGDSRDEQFF | NP95-73 |
| HIV 5 | CASSLWVTGGEQFF | NP89-213 |
| HIV 6 | CASSFSSGRPGELF | NP95-73 |
| HIV 7 | CASSLTVSSYNEQ | NP95-73 |
| HIV 8 | RCASSSGANV | NP95-73 |
| HIV 9 | FCASRFERELGQPQ | NP-94-34 |
| HIV 10 | LCSVVTGDGYTF | NP95-184-O |
| HIV 11 | CASSLVGLRGNTEA | NP89-213 |
| HIV 12 | CASSLASYTEA | NP94-34 |
| HIV-1 | HIV-2 | HIV-3 | HIV-4 | HIV-5 | HIV-6 | HIV-7 | HIV-8 | HIV-9 | HIV-10 | HIV-11 | HIV-12 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HIV-1 gp120 ( × 10–6) | 0.0024 | >1 | 0.014 | 0.016 | >1 | 0.018 | >1 | >1 | >1 | >1 | 0.10 | 0.055 |
| HIV-1 Pol ( × 10–6) | 0.017 | 0.012 | 0.0081 | >1 | >1 | >1 | >1 | 0.036 | >1 | 0.028 | 0.025 | 0.0085 |
| HIV-1 Gag P24 ( × 10–6) | 0.052 | >1 | >1 | >1 | >1 | >1 | >1 | 0.078 | 0.08 | >1 | >1 | >1 |
| HIV-1 Gag P17 ( × 10–6) | 0.022 | 0.018 | 0.010 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| HIV-1 Tat ( × 10–6) | >1 | >1 | 0.028 | >1 | >1 | >1 | 0.020 | 0.034 | 0.022 | >1 | >1 | 0.015 |
| HIV-1 Nef ( × 10–6) | 0.031 | >1 | >1 | >1 | 0.0037 | >1 | 0.011 | 0.033 | >1 | 0.024 | 0.072 | 0.0085 |
| CMV (AD169) Mab ( × 10–6) | >1 | >1 | >1 | |||||||||
| HCV Core ( × 10–6) | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
| RBT M. tuber. ( × 10–6) | 0.011 | >1 | >1 | >1 | >1 | 0.10 | >1 | >1 | 0.0085 | >0.1 | >1 | >1 |
| Anti-Mycob. ( × 10–6) | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Root-Bernstein, R. Human Immunodeficiency Virus Proteins Mimic Human T Cell Receptors Inducing Cross-Reactive Antibodies. Int. J. Mol. Sci. 2017, 18, 2091. https://doi.org/10.3390/ijms18102091
Root-Bernstein R. Human Immunodeficiency Virus Proteins Mimic Human T Cell Receptors Inducing Cross-Reactive Antibodies. International Journal of Molecular Sciences. 2017; 18(10):2091. https://doi.org/10.3390/ijms18102091
Chicago/Turabian StyleRoot-Bernstein, Robert. 2017. "Human Immunodeficiency Virus Proteins Mimic Human T Cell Receptors Inducing Cross-Reactive Antibodies" International Journal of Molecular Sciences 18, no. 10: 2091. https://doi.org/10.3390/ijms18102091
APA StyleRoot-Bernstein, R. (2017). Human Immunodeficiency Virus Proteins Mimic Human T Cell Receptors Inducing Cross-Reactive Antibodies. International Journal of Molecular Sciences, 18(10), 2091. https://doi.org/10.3390/ijms18102091